")
Back to Journals » International Journal of Nanomedicine » Volume 19
Staphylococcus Aureus Membrane Vesicles Kill Tumor Cells Through a Caspase-1-Dependent Pyroptosis Pathway
Authors Li M, Wang Y, Liu H, Huang X, Peng H, Yang Y, Hu Z, Dou J, Xiao C, Chen J , Shang W, Rao X
Received 15 January 2024
Accepted for publication 19 April 2024
Published 3 May 2024 Volume 2024:19 Pages 4007—4019
DOI https://doi.org/10.2147/IJN.S455158
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jie Huang
Video abstract presented by Li.
Views: 54
Mengyang Li,1,* Yuting Wang,2,* He Liu,2 Xiaonan Huang,2 Huagang Peng,2 Yi Yang,2 Zhen Hu,2 Jianxiong Dou,2 Chuan Xiao,2 Juan Chen,3 Weilong Shang,2 Xiancai Rao1,2
1Department of Microbiology, School of Medicine, Chongqing University, Chongqing, 400044, People’s Republic of China; 2Department of Microbiology, College of Basic Medical Sciences, Army Medical University, Key Laboratory of Microbial Engineering Under the Educational Committee in Chongqing, Chongqing, 400038, People’s Republic of China; 3Department of Pharmacy, The Second Affiliated Hospital, Army Medical University, Chongqing, 400037, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiancai Rao; Weilong Shang, Department of Microbiology, College of Basic Medical Sciences, Army Medical University, Key Laboratory of Microbial Engineering under the Educational Committee in Chongqing, Chongqing, 400038, People’s Republic of China, Email [email protected]; [email protected]
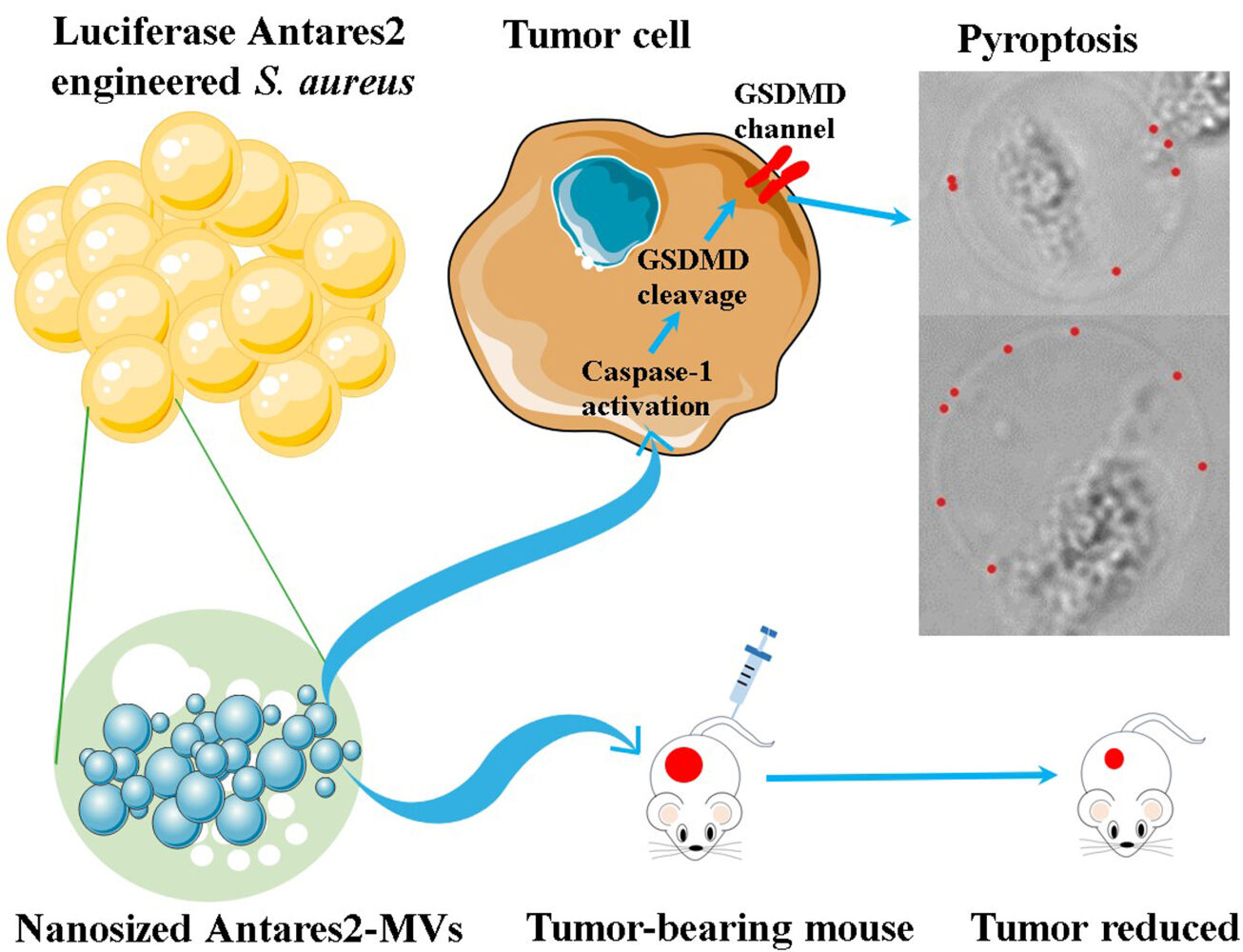
Introduction: Nanosized outer membrane vesicles (OMVs) from Gram-negative bacteria have attracted increasing interest because of their antitumor activity. However, the antitumor effects of MVs isolated from Gram-positive bacteria have rarely been investigated.
Methods: MVs of Staphylococcus aureus USA300 were prepared and their antitumor efficacy was evaluated using tumor-bearing mouse models. A gene knock-in assay was performed to generate luciferase Antares2–MVs for bioluminescent detection. Cell counting kit-8 and lactic dehydrogenase release assays were used to detect the toxicity of the MVs against tumor cells in vitro. Active caspase-1 and gasdermin D (GSDMD) levels were determined using Western blot, and the tumor inhibition ability of MVs was determined in B16F10 cells treated with a caspase-1 inhibitor.
Results: The vesicular particles of S. aureus USA300 MVs were 55.23 ± 8.17 nm in diameter, and 5 μg of MVs remarkably inhibited the growth of B16F10 melanoma in C57BL/6 mice and CT26 colon adenocarcinoma in BALB/c mice. The bioluminescent signals correlated well with the concentrations of the engineered Antares2–MVs (R2 = 0.999), and the sensitivity for bioluminescence imaging was 4 × 10− 3 μg. Antares2–MVs can directly target tumor tissues in vivo, and 20 μg/mL Antares2–MVs considerably reduced the growth of B16F10 and CT26 tumor cells, but not non-carcinomatous bEnd.3 cells. MV treatment substantially increased the level of active caspase-1, which processes GSDMD to trigger pyroptosis in tumor cells. Blocking caspase-1 activation with VX-765 significantly protected tumor cells from MV killing in vitro and in vivo.
Conclusion: S. aureus MVs can kill tumor cells by activating the pyroptosis pathway, and the induction of pyroptosis in tumor cells is a promising strategy for cancer treatment.
Keywords: Staphylococcus aureus, membrane vesicles, Antares2, cancer therapy, pyroptosis, caspase-1, GSDMD
Graphical Abstract:
Introduction
Cancer, including melanoma and colorectal carcinoma, is currently a serious health challenge worldwide.1–3 Timely surgery, chemotherapy, and radiotherapy are the main strategies for cancer treatment.4 To date, immunotherapy using RNA molecules, immunostimulatory agents, and immune-checkpoint inhibitors has become an increasingly common type of antitumor treatment.5–7 Membrane vesicles (MVs) are spherical, lipid-bilayer, and nanosized (20–400 nm) structures secreted by bacteria during their growth in vitro and in vivo.8 MVs were first observed in Escherichia coli about 50 years ago, and the MVs of Gram-negative bacteria are mainly released from the outer membrane, namely, outer membrane vesicles (OMVs).9,10 Owing to the carriage of bacterium-derived lipopolysaccharides (LPSs), phospholipids, proteins, and nucleic acids, several studies have demonstrated that OMVs have antitumor effects.11,12 OMVs produced by the E. coli strain ATCC 11775 exhibit unique antitumor efficiency in BALB/c nude mice with SK-N-SH neuroblastoma.13 Ren et al14 designed an ingenious antitumor nanoparticle by loading the Polybia–mastoparan I into E. coli (BL21) OMVs and found the chimeric peptide-encapsulated OMVs can suppress bladder cancer cells (MB49 and UMUC3) in vitro and in vivo. Therefore, bacterial MVs provide a novel avenue for clinical antitumor management.
The mechanisms underlying the killing of tumor cells by OMVs are complicated. The pathogen-associated molecular patterns (PAMPs) of OMV-loaded components can stimulate an antitumor immune response.15,16 Bacterial peptidoglycans and LPSs on the surfaces of OMVs can be recognized by a host’s Toll-like receptor (TLR) 2 and TLR4, respectively, and this process results in an inflammatory response.17 The intravenous injection of E. coli–derived OMVs can stimulate interferon γ- mediated antitumor immunoreaction in mice.11 By contrast, virulence factors, such as bacterial hemolysins, in MVs can damage mitochondrial function, activate caspase-3, and finally trigger cell apoptosis or pyroptosis.17,18 However, clarifying the specific molecules involved in the OMVs that exert diverse antitumor effects are quite difficult.
Gram-positive bacteria lack an outer membrane, and thus, the secretion of MVs by the bacteria had not been uncovered until 2009.19 The antitumor activity of Gram-positive bacterial MVs was seldom investigated. Jiang et al20 showed that MVs derived from Bifidobacterium can inhibit the growth of triple-negative breast cancer cells. Lentilactobacillus buchneri strain HBUM07105-produced MVs can reduce the viability of human gastric adenocarcinoma AGS cells and colorectal adenocarcinoma HT-29 cells in vitro.21 However, the mechanisms for Gram-positive bacterial MVs killing of tumor cells are unclear. Uncertain components in bacterial MVs may induce apoptosis in the tumor cells.20,21 The hemolysin-loaded pneumococcal MVs can be internalized by A549 lung adenocarcinoma cells, while the production of proinflammatory cytokines is irrespective of pneumolysin content.22 In this study, MVs of Staphylococcus aureus USA300 were prepared and characterized. Engineered S. aureus Antares2-MVs can directly target and inhibit xenograft tumors in mice. In vitro, S. aureus MVs decreased the viability of B16F10 and CT26 tumor cells, and increased active caspase-1 in B16F10 cell supernatants triggered pyroptosis in tumor cells after MV treatment, resulting in the cleavage of gasdermin D (GSDMD). Inhibition of caspase-1 activation rescued the death of tumor cells treated with S. aureus MVs in vitro, and reduced the antitumor activity of MVs against xenograft tumors in mice. Overall, this study verified the antitumor activity of S. aureus MVs and provided an insight into their antitumor mechanism, which may benefit the practice of cancer treatment.
Materials and Methods
Bacterial Strains and Cell Lines
S. aureus USA300 strain FPR3757 (GenBank accession no. CP000255.1) was kindly provided by Prof. Min Li (Shanghai Jiao Tong University). S. aureus RN4220 (NCTC 8325–4) was gifted by Prof. Baolin Sun (University of Science and Technology of China). Escherichia coli DH5α (TransGen, China) was cultured in Luria-Bertani (LB) broth or on LB agar (Oxoid, UK) at 37°C. S. aureus strains were cultured in brain heart infusion (BHI) (Oxoid, UK) at 37°C. When required, chloramphenicol (20 μg/mL) and ampicillin (100 μg/mL) were added. E. coli-S. aureus shuttle vector, pBT2 (from Baolin Sun) was used for bacterial engineering.
B16F10 murine melanoma cell line, CT26 murine colon adenocarcinoma cell line, and mouse brain microvascular endothelial cell line bEnd.3 were purchased from the American Type Culture Collection (ATCC, Shanghai). B16F10 and bEnd.3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA). CT26 cells were cultured in RPMI 1640 medium (Gibco). All media used for cell culture were supplemented with 10% (v/v) fetal bovine serum (Zeta Life, USA) and 1% (v/v) penicillin-streptomycin solution (Gibco). Cells were grown at 37°C incubator with a humidified atmosphere containing 5% (v/v) CO2.
MV Preparation
S. aureus MVs were prepared according to a previously established protocol.23 Briefly, overnight culture of S. aureus USA300 was 1:200 diluted and inoculated into fresh BHI medium (Oxoid), cultured for 20 h at 37°C with shaking (200 rpm). Then, the culture was centrifuged at 5000 ×g for 30 min at 4°C and filtered through a 0.45 μm sterile membrane filter (Millipore, Billerica, USA). Afterward, the filtrate was concentrated approximately 50-fold using a 100 kDa cut-off hollow fiber membrane column (Amersham Biosciences, Piscataway, NJ, USA). Bacterial cell debris in the supernatant was removed using a 0.22 μm sterile filter. Finally, S. aureus MVs were obtained after ultracentrifugation (150,000 ×g, 2.5 h, 4°C) using an ultracentrifuge (Himac, Japan), and the pellets were resuspended in sterile phosphate buffered saline (PBS, pH 7.2). The protein concentration of the MVs was measured using a Bradford Protein Concentration Assay Kit (Beyotime, China). The purified MVs were aliquoted and stored at −80°C until use.
Characterization of S. Aureus MVs
S. aureus MVs were observed with a transmission electron microscope (TEM), as previously described.23 Briefly, the purified MVs were placed on 230-mesh formvar/carbon-coated copper grids (Zhongjingkeji Technology, China) and negatively stained with 2% (w/v) uranyl acetate for 30 s. Electron micrographs of MVs were obtained using a JEM1011 microscope (JEOL, Japan).
The size of the purified MVs was determined by dynamic light scattering analysis using a Zetasizer Nano ZS90 (Malvern, UK). Briefly, 0.1 mg/mL of the purified S. aureus MVs were prepared in PBS. Reads of 60 s duration were performed in triplicates at 25°C. Data outputs were obtained using the Zetasizer (Nano, μV, APS) software version 7.13 (Malvern, UK). The average number of MV particles at the binned center in the output was adjusted by the dilution factor using the GraphPad Prism software 9.0.0.
Evaluation of Antitumor Effect of S. Aureus MVs in vivo
C57/BL6 or BALB/c female mice (6–8 weeks old) were purchased from the Vital River Laboratory Animal Technology (Beijing, China) and maintained in specific pathogen-free (SPF) animal laboratories. To determine the toxicity of S. aureus MVs, C57/BL6 mice were randomly divided into five groups (n = 10 per group) and intravenously challenged once every other day with 1, 5, 10, or 20 μg of MVs. Mice injected with PBS were used as the controls. The survival of mice was monitored daily for up to 12 d.
To evaluate the antitumor effect of S. aureus MVs in vivo, C57/BL6 or BALB/c mice were subcutaneously injected with 100 μL B16F10 melanoma or CT26 colon adenocarcinoma cells (2.5 × 106 cells per mouse) into the backs of depilated mice (n = 5 per group). After 24 h of tumor cell injection, mice were intravenously administered 100 μL of a solution containing 5 μg MVs once every other day, six times. Mice injected with PBS were used as the controls. Tumor size and weight were measured daily. Tumor volume was calculated as previously described,11,24 and a formula used was as the following:

Construction of Luciferase Antares2–MV-Produced S. Aureus Strain
S. aureus enolase (Eno) was selected as a target for luciferase fusion as previous described.23 The nucleotide sequence of luciferase Antares2 according the codon usage bias of S. aureus USA300 was chemically synthesized by BGI-Shenzhen (China) and cloned in the pUC-antares2 plasmid.25 The antares2 was amplified with polymerase chain reaction (PCR) and in-frame fused with the eno gene within the genome of S. aureus USA300 using homologous recombinant strategy.26 Briefly, the antares2 gene was amplified using primers eno-antares2-F3 (5′-cttagataaaatggtgagcaagggcgag-3′) and R3 (5′-gaaaactcgagtcacttgtacagctcgtccatgc-3′) from the pUC-antares2 plasmid. For site-specific homologous recombination, the left and right homologous arms across the stop codon of eno gene were designed and amplified from S. aureus USA300 genomic DNA using the primers up-eno-F3 (5′-gagctcggtacccgggatgatcgcattagacggt-3′)/R3(5′-tgctcaccattttatctaagttatagaatgatttgataccg-3′) and down-eno-F3 (5′-caagtgactcgagttttctttataatcaaatgctgac-3′)/R3 (5′-gataaactaccgcattactgcttttaccttcttggagtag-3′). The fusion fragment (left arm–antares2–right arm) was obtained by overlap PCR with the primer pair up-eno-F3/down-eno-R3 and cloned into the temperature-sensitive shuttle vector pBT2 using a one-step cloning strategy with a Gibson Assembly kit (New England Biolabs, Beijing, China). The ligates were transformed into E. coli DH5α cells to generate pBT2-antares2. The pBT2-antares2 plasmid was transformed into S. aureus RN4220 for restrictive modification, and the resulting plasmid was then transformed into USA300. The integration of the plasmid into bacterial chromosome was induced by cultivating plasmid-carrying USA300 at 42°C for 20 h, followed by cultivation at 25°C for 20 h to obtain plasmid missing mutants. The USA300/Eno–Antares2 strain was confirmed using PCR and DNA sequencing.
Western Blot Analysis
To detect the expression of the luciferase fusion protein Eno–Antares2 in the engineered USA300/Eno-Antares2 and its MVs, the prepared MVs and bacterial cell lysates were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (GE Healthcare). The PVDF membrane was blocked using blocking solution (Beyotime, China). The target proteins were probed with mouse anti-Eno polyclonal antibodies,23 followed by secondary sheep anti-mouse antibodies coupled with horseradish peroxidase (Abmart, China). Western blot images were obtained using the SuperSignal West Atto Substrate (Thermo Fisher Scientific, USA).
Bioluminescence Detection of Antares2–MVs in vitro
Antares2–MVs were prepared from the culture of engineered S. aureus USA300/ Eno–Antares2, as described above. For in vitro bioluminescence imaging, 50 μL of Antares2–MVs were diluted to various concentrations (2.5, 0.5, 0.1, 0.02, 0.004, 0.0008, and 0 μg/100 μL) in a black 96-well plate, and normal MVs (2.5 μg) were used as controls. The substrate hydrofurimazine (HFZ) of luciferase Antares2 was dissolved in a polyethylene glycol-300 (PEG-300) formulation containing 10% (v/v) glycerol, 10% ethanol (v/v), 10% (v/v) hydroxypropylcyclodextrin, and 35% (w/v) PEG-300.27 Equal volumes of the HFZ substrate (100 μM) were subsequently added and mixed well. The total photon flux (p/sec/cm²/sr, p/s) in each well was determined using an IVIS® Lumina LT Series III system (PerkinElmer, USA).
Bioluminescence Tracking of Antares2–MVs in vivo
C57BL/6 and BALB/c female mice were subcutaneously inoculated with B16F10 and CT26 tumor cells (2.5 × 106 cells per mouse). A total of 5 μg of Antares2–MVs were intravenously administered to each mouse (n = 3 per group). Injection of 100 μL of HFZ substrate (100 μM) around the tumor was performed 1, 3, 6, 12, and 24 h post-injection of MVs. Total photon flux and images were obtained using an IVIS® Lumina LT Series III system (Perkin Elmer, USA).
Direct Antitumor Activity of S. Aureus MVs in vitro
Cell counting kit-8 (CCK-8) and lactate dehydrogenase (LDH) release assays were performed to determine the cytotoxicity of S. aureus MVs against tumor and normal control cells in vitro. Experiments were conducted with protocols as previously described.18,28 As for CCK-8 assay, B16F10, CT26, and bEnd.3 cells were cultivated in 96-well plates until the cell density reached 70%–80%. Then, the medium was replaced with fresh medium (100 μL per well) supplemented with 10 μL of S. aureus MVs at final concentrations of 5, 20, and 100 μg/mL. The addition of 10 μL of PBS served as a control. Cells were incubated at 37°C for 1, 3, 6, 12, and 24 h. At each time point, 10 μL of CCK-8 reagent (ABclonal, China) was added to each well and incubated for another 1 h. The optical density (OD) at 450 nm was measured using a microplate reader (Thermo Fisher, USA). The cell viability was calculated according to the formula recommended in the manufacturer’s instructions. For the LDH cytotoxicity assay, cells of interest were seeded into 96-well plates and cultured in media containing MVs at final concentrations of 5, 20, and 100 μg/mL. PBS was used as a control. The cell mortality rates were assessed using an LDH cytotoxicity assay kit (Beyotime) after treatment with S. aureus MVs for 1, 3, 6, 12, and 24 h.
Caspase-1 Activity Assay
B16F10 cells were seeded into a 96-well plate with 2.0 × 104 cells and cultured at 37°C with 5% CO2. When the cell density reached 80%, 10 μL of S. aureus MVs diluted in DMEM complete medium was added at final concentrations of 5, 20, and 100 μg/mL. PBS was used as a control. After 8 h of incubation at 37°C, cell supernatants were analyzed using a kit of Caspase-Glo® 1 Inflammasome Assay (Promega) to detect caspase-1 activity, as described.18
Detection of Active Caspase-1 and Cleaved GSDMD
B16F10 cells were cultured in 96-well plates until the cell density reached 80% confluency. The original medium was then replaced with fresh medium supplemented with 20 μg/mL of S. aureus MVs or an equivalent volume of PBS. Then, the cells were incubated at 37°C for 6 h. For inhibition group, B16F10 cells were pretreated with 25% (w/v) VX-765 (MCE, USA) for 30 min to block MV-induced caspase-1 activation. To detect active caspase-1, cell supernatant was aspirated and centrifuged. To detect cleaved GSDMD, cell supernatant was precipitated with 10% (v/v) trichloroacetic acid (TCA) and incubated at 4°C overnight. After washing thrice with cold acetone, the samples were dried and resuspended in PBS. Adherent cells were lysed with 200 μL RIPA Lysis Buffer (Beyotime) supplemented with 1% (w/v) phenylmethanesulfonyl fluoride (PMSF) (Solarbio, China). Cell debris were cleared by centrifugation at 15,000 ×g for 10 min at 4°C. The concentration of the cell lysate was measured using the Bradford protein concentration assay kit (Beyotime). Western blot was performed as described previously.23 The primary antibodies used were mouse anti-pro Caspase-1 or GSDMD (1 μg/mL, ABcam). After incubation with horseradish peroxidase-conjugated secondary antibodies, immunoreactive bands were observed after the addition of SuperSignal West Atto Substrate (Thermo Fisher Scientific, USA). Cell viability and mortality rates were determined using the CCK-8 and LDH assays, respectively.
Caspase-1 Inhibition with VX-765 Attenuated the Antitumor Activity of S. Aureus MVs in vivo
To evaluate the effects of VX-765 on the antitumor activity S. aureus MVs in vivo, C57/BL6 mice were subcutaneously injected with 100 μL B16F10 melanoma cells (2.5 × 106 cells per mouse) into the backs of depilated mice (n = 5 per group). VX-765 solution was prepared as described above. After 24 h of melanoma cell injection, 50 mg/kg VX-765 solution or vehicle control were intraperitoneally injected as previously described,29 and the S. aureus USA300 MVs (5 μg per mouse) were intravenously administered 1 h postinjection of VX-765. The treatment was performed once every other day. The mouse weight, tumor size, and tumor weight were measured.
Statistical Analysis
All data were analyzed using GraphPad Prism Software, version 9.0.0. The experiments were conducted at least three times. Data are presented as the mean ± standard deviation (SD) or mean ± standard error of the mean (SEM). Survival analysis was performed using the log-rank test. The unpaired t-test was used to compare the two groups. One-way and two-way analyses of variance (ANOVA) were used for multiple group comparisons. A p value less than 0.05 was considered statistically significant difference.
Results
MVs Derived from S. Aureus USA300 Killed Tumors in vivo
Bacterial OMVs can effectively induce antitumor immune responses.30 To investigate the antitumor activity of MVs from Gram-positive bacteria, S. aureus USA300 was cultured, and its MVs were prepared (Figure 1A). The nanosized lipid-bilayer vesicular structures of S. aureus MVs were observed by TEM (Figure 1B), and the MVs presented an average diameter of 55.23 ± 8.17 nm (Figure 1C). Variable amounts of S. aureus MVs were intravenously administered to C57BL/6 mice to evaluate toxicity in vivo. The results showed that mouse survival rates were 0%, 40%, 100%, and 100% 12 d after intraperitoneal challenge with 20, 10, 5, and 1 μg of MVs, respectively (Figure 1D). Therefore, we selected 5 μg MVs as the treatment dose for subsequent animal experiments.
 |
Figure 1 Antitumor effect of S. aureus MVs in vivo. (A) Schematic diagram showing the procedure of S. aureus MV preparation. (B) TEM observation of the MVs produced by S. aureus USA300. The bar represents 100 nm. (C) Size distribution of USA300 MVs measured by a dynamic light scattering assay. (D) Survival rate of C57BL/6 mice after intravenously injected diverse concentrations of S. aureus MVs (n = 10 per group). PBS was used as control. (E) Schematic diagram showing the experimental design of antitumor effect of S. aureus MVs in vivo. C57BL/6 female mice were subcutaneously injected with B16F10 cells on day 0, and intravenously injected with 5 μg of bacterial MVs on day 1, and MV treatment was performed once every other day up to 11 days. (F) Tumor images and (G) weights harvested at day 11 posttreatment. (H) Tumor volumes of mice bearing B16F10 tumor measured after tumor formation. (I) Variation of body weight in tumor-bearing mice during MV-treatment. Survival analysis (D) was calculated by log-rank test. Data in G–I are presented as mean ± SD. Statistical significance was calculated via t-test or two-way ANOVA. **p < 0.01 and *p < 0.05. |
The antitumor effects of S. aureus MVs were evaluated in C57BL/6 mice bearing B16F10 melanoma and BALB/c mice carrying CT26 colon adenocarcinoma. Approximately 2.5 × 106 B16F10 or CT26 cells (100 μL) were subcutaneously injected into the back of each mouse on day 0 (n = 5) to generate tumor-bearing models, and 5 μg of S. aureus MVs were intravenously injected into the tumor-bearing mice once every other day (Figure 1E). After six treatments, the tumor volume and weight were significantly reduced compared to those in the PBS control (p < 0.01, Figure 1F–H and Supplementary Figure 1A–C). However, the body weights of the MV-treated C57BL/6 mice on day 11 notably decreased (Figure 1I), but not in BALB/c mice (Supplementary Figure 1D), which may be caused by the mouse sensibility to uncertain virulence factors packaged in the MVs.28 Collectively, these results suggest that MVs prepared from S. aureus USA300 have antitumor activity in vivo.
S. Aureus MVs Targeted Tumor Tissues in vivo
OMVs of Gram-negative bacteria can passively accumulate around tumor tissues through enhanced permeability and retention.11,31,32 However, fluorescent Cy7-labeled OMVs are difficult to detect in vivo. In the present study, a highly sensitive luciferase gene Antares227 was genetically in-frame fused with the 3ʹ-terminal of the eno gene of S. aureus USA300 (Figure 2A, Supplementary Figure 2A and B), which encodes enolase that can be incorporated into MVs.24 Western blot showed that the Eno–Antares2 fusion proteins were successfully loaded onto the MVs of S. aureus USA300 (Figure 2B and Supplementary Figure 2C), namely, Antares2–MVs. The addition of different substrates, such as HFZ, diphenylterazine (DTZ), and furimazine (FUR), produced macroscopically visible orange-red light in tubes containing Antares2–MVs but not normal MVs (Figure 2C). Antares2–MVs catalyzed HFZ to emit photons in a dose-dependent manner (Figure 2D). The total bioluminescence signals were highly correlated with the concentrations of Antares2–MVs (R2 = 0.999), which showed a sensitivity of 4 × 10−3 μg of Antares2–MVs detected via in vitro bioluminescence imaging (Figure 2D and E). These results indicate that the Antares2–MVs derived from engineered S. aureus USA300/Eno–Antares2 are sensitive for the in vivo monitoring of bacterial MVs.
 |
Figure 2 S. aureus MVs targeted tumor tissues in vivo. (A) Schematic diagram of the Eno-fused Antares2 luciferase in the engineered S. aureus USA300. (B) Western blot analysis of Eno–Antares2-loaded MVs. (C) Bioluminescence photographs of Antares2–MVs that catalyzed diverse substrates in tubes. (D) Bioluminescence signals of diverse amounts of Antares2–MVs (2.5, 0.5, 0.1, 0.02, 0.004, and 0.0008 μg) catalyzed HFZ in vitro. Normal MVs (2.5 μg) served as negative control. (E) Relationship between Antares2–MV amount and the total photon flux by simple linear regression (n = 3). Data are presented as mean ± SD. (F) Accumulation of MVs in tumor tissues. B16F10 tumor-bearing mice were intravenously injected with 5 μg of Antares2–MVs and 100 μmol HFZ substrate, and then the bioluminescence signals were detected at the time indicated. PBS was injected as the control. (G) Total photon flux in (F) changed over time. Data are presented as mean ± SEM. Abbreviations: HFZ, hydrofurimazine; DTZ, diphenylterazine; FUR, furimazine. |
B16F10 tumor-bearing mice (n = 3 per group) were generated, and 5 μg of Antares2–MVs was intravenously injected into each mouse to trace the MV distribution after administration of 100 μL of HFZ substrate. The Antares2–MVs could accumulate in tumor tissues (Figure 2F and Supplementary Figure 3A). The bioluminescence intensity decreased over time, and detectable signals appeared within 12 h postinjection of Antares2–MVs and HFZ substrate (Figure 2G and Supplementary Figure 3B). Collectively, these findings suggest that S. aureus MVs can directly target tumor tissues in vivo.
S. Aureus MVs Killed Tumor Cells in vitro
To determine whether the observed S. aureus MVs in the tumor tissues in vivo could directly kill tumor cells, we treated melanoma B16F10 and colon adenocarcinoma CT26 cells with various concentrations of MVs at different times (Supplementary Figure 4). The CCK-8 assay revealed that the viability of tumor cells decreased considerably after treatment with S. aureus MVs at concentrations over 20 μg/mL (Figure 3A and B). S. aureus MVs can load a variety of pore-forming toxins, such as alpha-hemolysin and leukocidin-like proteins, which may play a role in cytotoxicity.21 LDH release assays were performed to evaluate the toxicity of S. aureus MVs against tumor cells. The results showed that LDH release increased in a dose-dependent manner after treatment with different concentrations of the MVs (Figure 3C and D). However, the non-carcinomatous cell line bEnd.3, which was generated from mouse brain microvascular endothelial cells,14 was resistant to MV killing at different times after treatment with 20 μg/mL MV (Figures 3E, F, and Supplementary Figure 4C). These data indicate that S. aureus MVs can directly kill tumor cells, and that their antitumor activity is specific.
 |
Figure 3 S. aureus MVs killed tumor cells in vitro. The viability rates of B16F10 (A) and CT26 (B) cells. Tumor cells were treated with various concentrations of MVs (5, 20, and 100 μg/mL) for 1, 3, 6, 12, and 24 h, respectively (n = 3 for each). PBS treatment served as controls. Cell viability was measured by CCK-8 assay. The mortality rates of B16F10 (C) and CT26 (D) cells. Tumor cells were treated as described above (n = 3 for each). Cell mortality was measured by LDH cytotoxicity assay. Cells were treated with 20 μg/mL of MVs for 1, 3, 6, 12, and 24 h, the cell viability rates (E) and cell mortality rates (F) of tumor cells B16F10 and CT26, and the non-carcinomatous bEnd.3 cells measured by using CCK-8 and LDH cytotoxicity assays, respectively (n = 3). Data are presented as mean ± SD. Statistical significance was calculated via one-way or two-way ANOVA. ***p < 0.001, **p < 0.01, and *p < 0.05. |
S. Aureus MVs Inactivated Tumor Cells Through the Pyroptosis Pathway
S. aureus MVs can induce pyroptosis in human macrophage THP-1 cells.18 To test whether S. aureus MVs can induce pyroptosis in tumor cells, we treated B16F10 cells with increasing concentrations of S. aureus MVs for 6 h and observed pyrogenic cells (Figure 4A and Supplementary Figure 5A). Then, caspase-1 activation in B16F10 cells after MV treatment was detected using the Caspase-Glo 1 inflammasome assay. The results demonstrated that caspase-1 activity significantly increased after treatment with 20 μg/mL MVs (Figure 4B and Supplementary Figure 5B). Western blot revealed similar caspase-1 levels in the lysates of B16F10 cells treated with S. aureus MVs and PBS (Figure 4C). However, increased levels of active caspase-1 were detected in the culture supernatant of B16F10 cells treated with MVs (Figure 4D). GSDMD is a generic substrate for caspase-1, which cleaves GSDMD to release the active gasdermin-N domain.33 Western blot demonstrated that the N-terminal GSDMD increased in the culture supernatants of B16F10 after treatment with S. aureus MVs (Figure 4D). Inhibition of caspase-1 activation by VX-765 markedly reduced the release of N-terminal GSDMD and active caspase-1 in B16F10 cells treated with MVs (Figure 4D).
 |
Figure 4 The antitumor activity of S. aureus MVs depended on caspase-1 activation. (A) The pyrogenic cells observed after treatment with S. aureus MVs. B16F10 cells were treated with 20 µg/mL of S. aureus MVs for 24 h, and a representative photograph was shown. (B) Detection of Caspase-1 activity. B16F10 cells were incubated with 0, 5, 20, and 100 µg/mL of S. aureus MVs for 6 h, and the active caspase-1 level in the culture supernatant was detected with a kit of Caspase-Glo® 1 Inflammasome Assay. The total flux of bioluminescence signals was determined (n = 3). Western blot analysis of (C) caspase-1 and GSDMD in the cell lysates and (D) active caspase-1 and cleaved N-terminal GSDMD in the culture supernatant. (E) CCK-8 assay and (F) LDH release assay. B16F10 cells were pretreated with VX-765 for 30 min to block MV-induced caspase-1 activation. The viability or mortality rates of B16F10 cells after treatment with 20 μg/mL of S. aureus MVs for 6 h were measured (n = 5). Data in (B), (E), and (F) are presented as the mean ± SD. Statistical significance was calculated by one-way ANOVA or t-test. ***p < 0.001 and *p < 0.05. |
Consistent with the observed release of active caspase-1 and N-terminal GSDMD, addition of the VX-765 inhibitor considerably increased the viability of B16F10 cells treated with S. aureus MVs (Figure 4E). Moreover, the mortality rate of B16F10 cells decreased significantly after treatment with the VX-765 inhibitor (Figure 4F). Overall, these data suggest that S. aureus MVs can activate caspase-1, which cleaves GSDMD and triggers pyroptosis in tumor cells.
Caspase-1 Inhibition Reduced the Antitumor Effect of S. Aureus MVs in vivo
To further investigate whether caspase-1 inhibition could affect the antitumor efficiency of S. aureus MVs in vivo, C57BL/6 mice (n = 5 per group) bearing B16F10 melanoma were generated. After 24 h of tumor cell injection, VX-765 (50 mg/kg) was intraperitoneally injected 1 h before MV treatment (5 μg per mouse), and six treatments with VX-765/MVs were provided. The results showed that the tumor volumes and weights in MVs and MVs+vehicle groups were comparable (Figure 5). However, VX-765 pretreatment remarkably weakened the antitumor activity of MVs (Figure 5B and D), although negligible variations in mouse weights were presented (Figure 5E). Taken together, VX-765 could inhibit the antitumor effect of MVs in vivo, and killing tumors by S. aureus MVs was caspase-1-dependent.
 |
Figure 5 VX-765 reduced the antitumor effect of MVs in vivo. (A) Representative xenograft tumors in mice taken at day 4, 6, 8, and 10 after treatment with S. aureus MVs and VX-765. (B) Tumor volumes and (C) sizes harvested at day 11 posttreatment. (D) Tumor weights of mice bearing B16F10 melanoma measured after tumor formation. (E) Variation of mouse weights during treatment. Data are presented as mean ± SD. Statistical significance was calculated via one-way or two-way ANOVA. ***p < 0.001, **p < 0.01, and ns indicates no significance. |
Discussion
MV-loaded components, including lipids, LPSs, proteins, and nucleic acids, are host bacterium dependent.23,30,34 MVs derived from diverse bacteria or strains may have distinct bioactivity.34 To obtain S. aurues MVs with antitumor potential, we used a hypervirulent USA300 strain isolated in 2000 and has undergone rapid clonal expansion worldwide.35 The purified MVs from S. aurues USA300 were 55.23 ± 8.17 nm in diameter, consistent with previous studies.23,36 We confirmed that 5 μg of USA300-derived MVs is safe for C57BL/6 mice. However, the survival rates of the MV-challenged mice decreased with increasing MV dose, and 20 μg of USA300 MVs killed all tested mice within 24 h post-injection (Figure 1D). Some toxic components can be enriched in MVs during formation and are the chief contributors to MV toxicity.23,28,37 However, the identification of the actual factors that play vital roles in USA300 MV toxicity is important, and we will investigate them in the future.
OMVs derived from several Gram-negative bacteria, such as E. coli, S. Typhimurium, and Porphyromonas gingivalis have antitumor potential.38 In this study, C57BL/6 mice were subcutaneously transplanted with CT26 colon adenocarcinoma or B16F10 melanoma cells, and 5 μg of MVs was intravenously injected once every other day to test the antitumor effects of MVs prepared from Gram-positive S. aureus. The results showed that the USA300-MVs remarkably suppressed the growth of transplanted carcinomas in mice (Figure 1F–H). Kim et al11 labeled OMVs with the fluorescent dye Cy7 and observed the accumulation of Cy7-OMVs in tumor tissues after systemic administration. Targeting tumor sites by nanoscale OMVs is passive.24,31,32 Encouraged by the high sensitivity of bioluminescence imaging, we engineered S. aureus USA300 by fusing the luciferase antares2 gene with chromosomal eno, which encodes Eno that can be enriched in MVs.23,39,40 Notably, bioluminescence signals were linearly associated with Antares2–MV doses (R2 = 0.999), and a sensitivity of 4 × 10−3 μg of MVs was achieved in vitro (Figure 2D and E). We further confirmed that Antares2–MVs could directly target tumor sites in mice (Figures 2F and Supplementary Figure 3A). This finding suggests that S. aureus MVs may exert direct antitumor activity. Accordingly, the direct antitumor effects of Antares2–MVs were observed in vitro, where the MV-induced immune function was excluded. The addition of 20 μg/mL Antares2–MVs significantly reduced the viability of B16F10 and CT26 tumor cells, but not non-carcinomatous bEnd.3 cells (Figure 3). These data are consistent with the findings of a recent study, in which E. coli OMVs engineered using Polybia–mastoparan I fusion peptides presented selective tumor-killing activity.14 S. aureus MVs can package an array of prototypic pore-forming toxins, including alpha-hemolysin, delta-hemolysin, and leukocidins.28,36 However, the selective cytotoxicity of S. aureus MVs against tumor cells is unclear. One possible explanation is that the expression levels of toxin receptors vary between tumorous and non-carcinomatous cells; however, this explanation requires further investigation.
Many mechanisms are associated with bacterial OMV-mediated tumor killing.13,18,41 Intumescent cells were observed in B16F10 cells after S. aureus MV treatment (Figure 4A and Supplementary Figure 5A), indicating that pyroptotic cell death occurred.41 Caspase-1 activity was considerably increased in the supernatant of B16F10 cells after treatment with 20 μg/mL S. aureus MVs, which is consistent with the results of Western blot (Figure 4D). Caspase-1 specifically cleaves the linker between the N- and C-terminal domains in GSDMD, and the released N-terminal GSDMD is a key element in triggering pyroptosis.33 Cleaved N-terminal GSDMD increased in the B16F10 culture supernatant. Inhibition of caspase-1 activation by VX-765 significantly inhibited MV-induced N-terminal GSDMD release (Figure 4D), cell death (Figure 4E and F), and in vivo antitumor activity (Figure 5A–D). Overall, these findings suggest that the caspase-1-dependent pyroptosis pathway is involved in tumor killing by S. aureus MVs.
Conclusions
In conclusion, our study focused on the antitumor potential of S. aureus MVs. By using engineered Antares2–MVs, we demonstrated that S. aureus MVs could directly target and accumulate at tumor sites in vivo. The direct antitumor effect of S. aureus MVs has been confirmed in melanoma and colon adenocarcinoma cells in vitro, and the antitumor activity of S. aureus MVs is dependent on the caspase-1-mediated pyroptosis pathway. These data provide novel insights into the antitumor potential of MVs derived from Gram-positive bacteria.
Ethics Approval
All animal experiments were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of the People’s Republic of China, and animal procedures were reviewed and approved by the Animal Use and Care Administrative Advisory Committee of the Army Medical University (protocol no. AMUWEC2020735).
Author Contributions
Professor Xiancai Rao and Doctor Weilong Shang share co-corresponding authorship. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82071857 to XR and 82072238 to WS).
Disclosure
All authors declare no competing of interests in this work.
References
1. Negura I, Pavel-Tanasa M, Danciu M. Regulatory T cells in gastric cancer: key controllers from pathogenesis to therapy. Cancer Treat Rev. 2023;120:102629. doi:10.1016/j.ctrv.2023.102629
2. Grafanaki K, Grammatikakis I, Ghosh A, et al. Noncoding RNA circuitry in melanoma onset, plasticity, and therapeutic response. Pharmacol Ther. 2023;248:108466. doi:10.1016/j.pharmthera.2023.108466
3. Rudnicki Y, Stapleton SM, Batra R, et al. Perianal Paget’s-an aggressive disease. Colorectal Dis. 2023;25(6):1213–1221. doi:10.1111/codi.16549
4. Jia Y, Wang X, Deng Y, et al. Pyroptosis provides new strategies for the treatment of cancer. J Cancer. 2023;14(1):140–151. doi:10.7150/jca.77965
5. Caproni E, Corbellari R, Tomasi M, et al. Anti-tumor efficacy of in situ vaccination using bacterial outer membrane vesicles. Cancers. 2023;15(13):3328. doi:10.3390/cancers15133328
6. Lucas MW, Versluis JM, Rozeman EA, Blank CU. Personalizing neoadjuvant immune-checkpoint inhibition in patients with melanoma. Nat Rev Clin Oncol. 2023;20(6):408–422. doi:10.1038/s41571-023-00760-3
7. Zhang Z, He C, Chen X. Designing hydrogels for immunomodulation in cancer therapy and regenerative medicine. Adv Mater. 2023;1:e2308894. doi:10.1002/adma.202308894
8. Toyofuku M, Nomura N, Eberl L. Types and origins of bacterial membrane vesicles. Nat Rev Microbiol. 2019;17(1):13–24. doi:10.1038/s41579-018-0112-2
9. Knox KW, Vesk M, Work E. Relation between excreted lipopolysaccharide complexes and surface structures of a lysine-limited culture of Escherichia coli. J Bacteriol. 1966;92(4):1206–1217. doi:10.1128/jb.92.4.1206-1217.1966
10. Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–184. doi:10.1146/annurev.micro.091208.073413
11. Kim OY, Park HT, Dinh NTH, et al. Bacterial outer membrane vesicles suppress tumor by interferon-gamma-mediated antitumor response. Nat Commun. 2017;8(1):626. doi:10.1038/s41467-017-00729-8
12. Jiang S, Fu W, Wang S, et al. Bacterial outer membrane vesicles loaded with perhexiline suppress tumor development by regulating tumor-associated macrophages repolarization in a synergistic way. Int J Mol Sci. 2023;24(13):11222. doi:10.3390/ijms241311222
13. Jin L, Zhang Z, Tan X, et al. Antitumor effect of Escherichia coli-derived outer membrane vesicles on neuroblastoma in vitro and in vivo. Acta Biochim Biophys Sin. 2022;54(9):1301–1313. doi:10.3724/abbs.2022127
14. Ren C, Li Y, Cong Z, et al. Bioengineered bacterial outer membrane vesicles encapsulated Polybia-mastoparan I fusion peptide as a promising nanoplatform for bladder cancer immune-modulatory chemotherapy. Front Immunol. 2023;14:1129771. doi:10.3389/fimmu.2023.1129771
15. Li M, Zhou H, Yang C, et al. Bacterial outer membrane vesicles as a platform for biomedical applications: an update. J Control Release. 2020;323:253–268. doi:10.1016/j.jconrel.2020.04.031
16. Briaud P, Carroll RK. Extracellular vesicle biogenesis and functions in Gram-positive bacteria. Infect Immun. 2020;88. doi:10.1128/IAI.00433-20
17. Long Q, Zheng P, Zheng X, et al. Engineered bacterial membrane vesicles are promising carriers for vaccine design and tumor immunotherapy. Adv Drug Deliv Rev. 2022;186:114321. doi:10.1016/j.addr.2022.114321
18. Wang X, Eagen WJ, Lee JC. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci USA. 2020;117(6):3174–3184. doi:10.1073/pnas.1915829117
19. Lee EY, Choi DY, Kim DK, et al. Gram-positive bacteria produce membrane vesicles: proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics. 2009;9(24):5425–5436.
20. Jiang Y, Wang L, Yang B, et al. Bifidobacterium-derived membrane vesicles inhibit triple-negative breast cancer growth by inducing tumor cell apoptosis. Mol Biol Rep. 2023;50(9):7547–7556. doi:10.1007/s11033-023-08702-z
21. Abedi A, Tafvizi F, Jafari P, Akbari N. The inhibition effects of Lentilactobacillus buchneri-derived membrane vesicles on AGS and HT-29 cancer cells by inducing cell apoptosis. Sci Rep. 2024;14(1):3100. doi:10.1038/s41598-024-53773-y
22. Codemo M, Muschiol S, Iovino F, et al. Immunomodulatory effects of pneumococcal extracellular vesicles on cellular and humoral host defenses. mBio. 2018;9(2):e00559–18. doi:10.1128/mBio.00559-18
23. Yuan J, Yang J, Hu Z, et al. Safe staphylococcal platform for the development of multivalent nanoscale vesicles against viral infections. Nano Lett. 2018;18(2):725–733. doi:10.1021/acs.nanolett.7b03893
24. Qing S, Lyu C, Zhu L, et al. Biomineralized bacterial outer membrane vesicles potentiate safe and efficient tumor microenvironment reprogramming for anticancer therapy. Adv Mater. 2020;32(47):e2002085.
25. Shang W, Hu Z, Li M, et al. Optimizing a high‐sensitivity NanoLuc‐based bioluminescence system for in vivo evaluation of antimicrobial treatment. mLife. 2023. doi:10.1002/mlf2.12091
26. Shang W, Rao Y, Zheng Y, et al. beta-Lactam antibiotics enhance the pathogenicity of methicillin-resistant Staphylococcus aureus via SarA-controlled lipoprotein-like cluster expression. mBio. 2019;10(3):e00880–19. doi:10.1128/mBio.00880-19
27. Su Y, Walker JR, Park Y, et al. Novel NanoLuc substrates enable bright two-population bioluminescence imaging in animals. Nat Methods. 2020;17(8):852–860. doi:10.1038/s41592-020-0889-6
28. Chen J, Lv Y, Shang W, et al. Loaded delta-hemolysin shapes the properties of Staphylococcus aureus membrane vesicles. Front Microbiol. 2023;14:1254367. doi:10.3389/fmicb.2023.1254367
29. Li Y, Shen Y, Jin K, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. 2019;30(3):477–492.e6. doi:10.1016/j.cmet.2019.06.016
30. Hosseini-Giv N, Basas A, Hicks C, et al. Bacterial extracellular vesicles and their novel therapeutic applications in health and cancer. Front Cell Infect Microbiol. 2022;12:962216. doi:10.3389/fcimb.2022.962216
31. Milling L, Zhang Y, Irvine DJ. Delivering safer immunotherapies for cancer. Adv Drug Deliv Rev. 2017;114:79–101. doi:10.1016/j.addr.2017.05.011
32. Aytar Celik P, Derkus B, Erdogan K, et al. Bacterial membrane vesicle functions, laboratory methods, and applications. Biotechnol Adv. 2022;54:107869. doi:10.1016/j.biotechadv.2021.107869
33. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
34. Jan AT. Outer membrane vesicles (OMVs) of Gram-negative bacteria: a perspective update. Front Microbiol. 2017;8:1053. doi:10.3389/fmicb.2017.01053
35. Diep BA, Gill SR, Chang RF, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–739. doi:10.1016/S0140-6736(06)68231-7
36. Fan R, Zhou Y, Chen X, et al. Porphyromonas gingivalis outer membrane vesicles promote apoptosis via msRNA-regulated DNA methylation in periodontitis. Microbiol Spectr. 2023;11(1):e0328822. doi:10.1128/spectrum.03288-22
37. Rivera J, Cordero RJB, Nakouzi AS, et al. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc Natl Acad Sci USA. 2010;107(44):19002–19007. doi:10.1073/pnas.1008843107
38. Hikita T, Miyata M, Watanabe R, Oneyama C. Sensitive and rapid quantification of exosomes by fusing luciferase to exosome marker proteins. Sci Rep. 2018;8(1):14035. doi:10.1038/s41598-018-32535-7
39. Hikita T, Miyata M, Watanabe R, Oneyama C. In vivo imaging of long-term accumulation of cancer-derived exosomes using a BRET-based reporter. Sci Rep. 2020;10(1):16616. doi:10.1038/s41598-020-73580-5
40. Wang X, Thompson CD, Weidenmaier C, Lee JC. Release of Staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat Commun. 2018;9(1):1379. doi:10.1038/s41467-018-03847-z
41. Munoz-Planillo R, Kuffa P, Martinez-Colon G, et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.