")
Back to Journals » International Medical Case Reports Journal » Volume 15
Corneal Refractive Surgery Considerations in Patients with Cystic Fibrosis and Cystic Fibrosis Transmembrane Conductance Regulator-Related Disorders
Authors Moshirfar M , Brown AH , Sulit CA , Corbin WM , Ronquillo YC , Hoopes PC
Received 6 July 2022
Accepted for publication 14 October 2022
Published 9 November 2022 Volume 2022:15 Pages 647—656
DOI https://doi.org/10.2147/IMCRJ.S381078
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Majid Moshirfar,1– 3 Alex H Brown,4 Christian A Sulit,4 Wyatt M Corbin,5 Yasmyne C Ronquillo,1 Phillip C Hoopes1
1Hoopes Vision Research Center, Hoopes Vision, Draper, UT, USA; 2John A. Moran Eye Center, University of Utah School of Medicine, Salt Lake City, UT, USA; 3Utah Lions Eye Bank, Murray, UT, USA; 4University of Arizona College of Medicine – Phoenix, Phoenix, AZ, USA; 5Loyola University Chicago Stritch School of Medicine, Maywood, IL, USA
Correspondence: Majid Moshirfar, Hoopes Vision Research Center, 11820 S. State St. #200, Draper, UT, 84020, USA, Tel +1 801-568-0200, Fax +1 801-563-0200, Email [email protected]
Abstract: This article discusses common ocular manifestations of cystic fibrosis (CF) and cystic fibrosis transmembrane conductance regulator-related disorders (CFTR-RD). A structured approach for assessing and treating patients with CF/CFTR-RD seeking corneal refractive surgery is proposed, as well as a novel surgical risk scoring system. We also report two patients with various manifestations of CFTR dysfunction who presented for refractive surgery and the outcomes of the procedures. Surgeons seeking to perform refractive surgery on patients with CF/CFTR-RD should be aware of mild to severe clinical manifestations of CFTR dysfunction. Specific systemic and ocular manifestations of CF include chronic obstructive pulmonary disease (COPD), bronchiectasis, recurrent pulmonary infections, CF-related diabetes and liver disease, pancreatic insufficiency, conjunctival xerosis, night blindness, meibomian gland dysfunction (MGD), and blepharitis. Corneal manifestations include dry eye disease (DED), punctate keratitis (PK), filamentary keratitis (FK), xerophthalmia, and decreased endothelial cell density and central corneal thickness. Utilization of the appropriate review of systems (ROS) and screening tests will assist in determining if the patient is a suitable candidate for refractive surgery, as CF/CFTR-RD can impact the health of the cornea. Collaboration with other medical professionals who care for these patients is encouraged to ensure that their CF/CFTR-RD symptoms are best controlled via systemic and other treatment options. This will assist in reducing the severity of their ocular manifestations before and after surgery.
Keywords: LASIK, PRK, cystic fibrosis, CF, cystic fibrosis transmembrane conductance regulator, CFTR, cystic fibrosis transmembrane conductance regulator-related disorder, CFTR-RD, cornea, refractive surgery
Introduction
Cystic Fibrosis (CF) is an autosomal recessive monogenic disorder with an incidence in the USA of approximately 1/4000 births. The incidence varies greatly between ethnic groups and is most prevalent in individuals of European descent.1 In the past, those diagnosed with CF rarely lived beyond childhood. However, advances in disease surveillance and treatments have significantly increased these individuals’ life expectancy, and those born with CF in the present day are predicted to live into their 4th or 5th decade.2 These patients may present to your clinic to be evaluated for corneal refractive surgery.
CF is caused by pathogenic mutations in both alleles of the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7, which produces the CFTR protein located on apical epithelial cell membranes of various organ systems, including the lungs, GI tract, kidneys, pancreas, male and female reproductive systems, and skin.3 It is also expressed in conjunctival epithelium, corneal epithelium, and corneal endothelium.4 The CFTR gene variants can be categorized into several classes depending on the mutation’s effect on the CFTR protein.5 The most common mutation, a class II homozygous deletion of phenylalanine 508 (F508del), impairs CFTR protein folding, processing, and function. More than 2000 mutations within the CFTR gene have been identified.6 Interestingly, some of these CFTR mutations are not associated with disease and do not appear to impact CFTR protein function.7 In all classes of CF-causing CFTR mutations, CFTR proteins cannot adequately pump chloride ions out of epithelial cells into the extracellular space. This disrupts ionic and osmolar gradients and causes hyperviscous mucus formation and decreased surface membrane hydration. This general mechanism underlies the multisystem effects observed in patients with CF.3
Although certain CFTR gene variants may correlate with specific pathologic manifestations, they do not necessarily correlate with clinical severity.5,8 Clinical severity may vary due to the combination of specific gene variants, the presence of modifier genes, environmental influences, and other factors.9 Thus, patients with CF can have a large variance in clinical presentation, disease severity, and progression. They can also be diagnosed between birth and late adulthood. Patients who do not meet the diagnostic criteria for CF but present with milder clinical symptoms may instead be diagnosed with CFTR-related disorder (CFTR-RD).10 These patients may have single organ system involvement, intermediate sweat chloride test measurements, and two or fewer CFTR mutations, one of which is not categorized as a CF-causing mutation.7,11,12 The age of symptom onset and diagnosis of CFTR-RD are generally later than patients with a classic form of CF.13 It is important to note that half of the individuals with CF aged 18 years or older have only mildly reduced lung function, with a quarter having moderately to severely reduced function.14
This article discusses common ocular manifestations of CF/CFTR-RD and proposes a structured approach for assessing and treating such patients seeking refractive surgery (Figure 1), as well as a novel surgical risk scoring system (Table 1). We also report two patients with CF and CFTR-RD, respectively, who presented for refractive surgery and the outcomes of the procedures. This article aims to assist eye care specialists in the care of this population. While this article focuses on patients with CF, the pathophysiology behind CFTR-related disorders is driven by CFTR dysfunction. Therefore, similar assessment and treatment strategies may be applied to these patient populations as well.
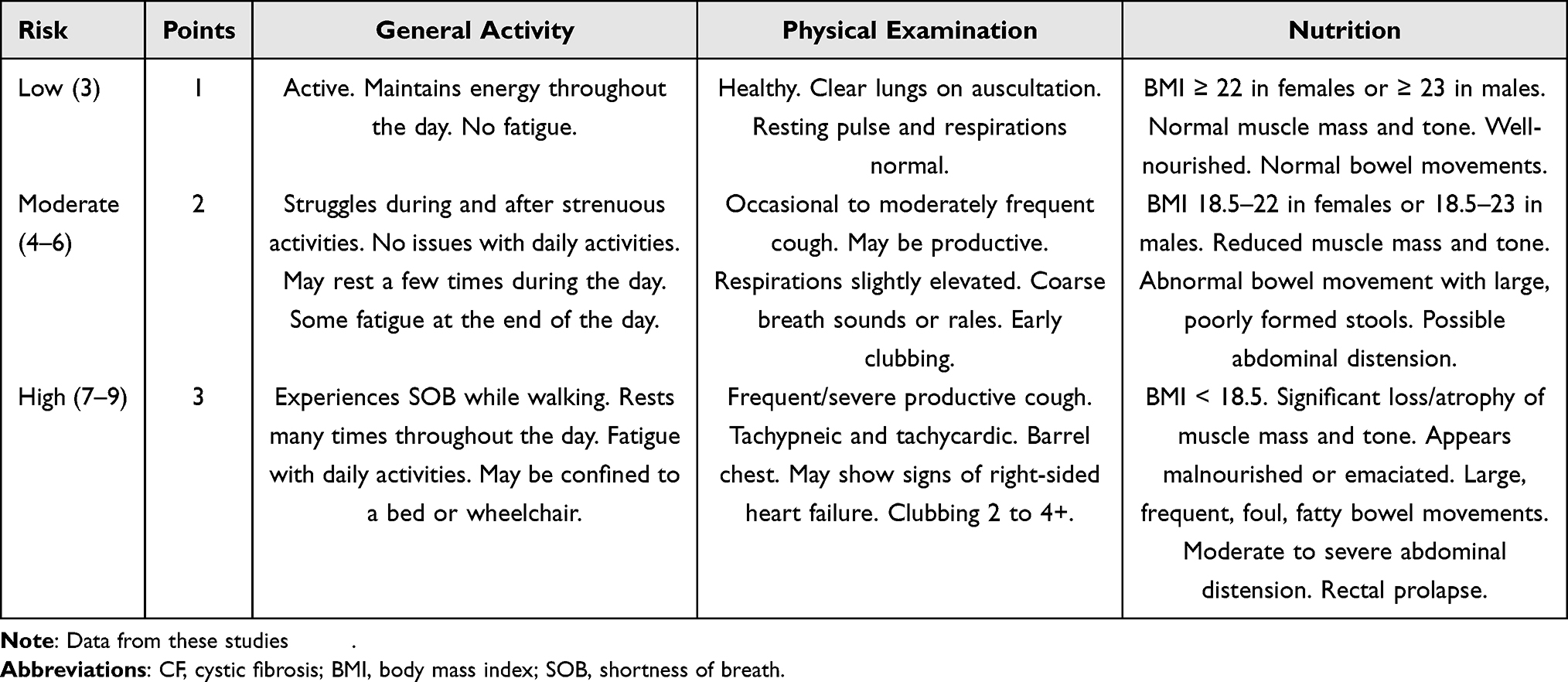 |
Table 1 Risk Stratification and CF Assessment Scoring Table for Corneal Refractive Surgery |
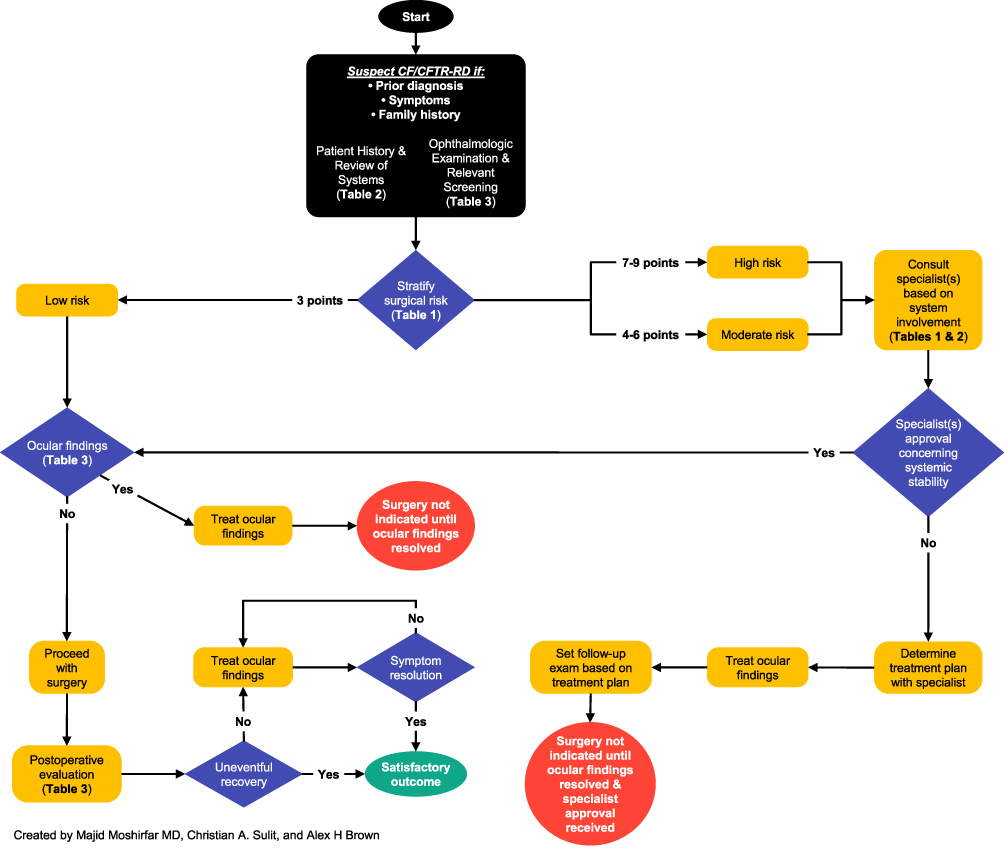 |
Figure 1 Sample structured approach to assessing and treating patients with CF/CFTR-RD before refractive surgery. Abbreviations: CF, cystic fibrosis. CFTR-RD, cystic fibrosis transmembrane conductance regulator-related disorder. Note: Data from Shwachman H, Kulczycki LL43. |
Cases
Patient #1
A 39-year-old Caucasian female with a history of CF presented for a LASIK consultation. She was a very healthy marathon runner who was well-versed in her condition and its management. She had a history of dry eye disease (DED) and minor CF-related pulmonary issues, specifically exercise-induced bronchospasms. She was taking Kalydeco® (Vertex Pharmaceuticals Incorporated, Boston, MA, USA) 150 mg tablets PO BID and using inhaled and nebulized medications. Review of systems (ROS) was unremarkable for skin abnormalities, pancreatic insufficiency, vitamin A deficiency, hepatic issues, GI issues, and COPD. Manifest refraction was −6.25 D OD and −5.75 D OS with a mean corneal thickness of 540 μm OD and 535 μm OS. Upon examination, it was discovered that this patient had conjunctival staining and superficial punctate keratopathy. Her Schirmer’s test was above 10 mm in 5 minutes and her tear break-up time (TBUT) was greater than 7 seconds. Refractive surgery was deferred. Treatment of DED consisted of punctal plugs and Refresh Plus Preservative Free (PF) Lubricant Eye Drops (AbbVie Inc., North Chicago, IL, USA). She showed significant clinical improvement after 6 months. Corneal staining was completely resolved but there was some conjunctival staining. She was started on Restasis® (AbbVie Inc., North Chicago, IL, USA) BID. She presented twice to the clinic with filamentary keratitis (FK) between 7–18 months after the initial visit, during which several corneal filaments were debrided using Jeweler’s forceps. She was cleared for refractive surgery 12 months after starting Restasis®. Due to concern for a high risk of postoperative ocular surface dryness, PRK was selected as the refractive surgery of preference for this patient.
Alcohol-assisted PRK monovision surgery was performed using an EX500 Wavelight Excimer Laser (Alcon Laboratories, Inc., Fort Worth, TX) using a 6.5 mm ablation zone with a transitional zone of 9.0 mm. The right eye was corrected for distance vision, and the left eye was under-corrected by 0.5 D. Mitomycin C was not used to avoid any risk of prolonged postoperative epithelial healing. An ACUVUE OASYS® bandage contact lens (Johnson & Johnson Vision Care, Inc., Jacksonville, FL) with a base curvature of 8.4 was applied postoperatively and removed after 5 days without any epithelial complications. The patient continued aggressive artificial tear treatment, topical moxifloxacin 0.5% QID OU for 8 days, and prednisolone acetate 1% OU QID for 4 weeks. Restasis® was restarted 8 days postoperatively after moxifloxacin was discontinued. Following 4 weeks of prednisolone acetate therapy, fluorometholone 0.1% was prescribed OU with a tapering regimen of TID for 3 weeks, BID for 3 weeks, and QD for 3 weeks.
During the first 6 months postoperatively, she presented twice to the clinic with FK. Several filaments were debrided from each eye using Jeweler’s forceps. The patient was placed on 10% topical N-acetylcysteine made by a compounding pharmacy to be used QID for 2 months, BID for 2 months, and was discontinued after. Treatment resulted in the complete eradication of FK. The patient’s postoperative refractive error was −0.25 D −0.5 D ×170° OD with UDVA of 20/25, and −0.75 D −0.75 D ×10° OS with UDVA of 20/50, correctable to 20/20 binocular vision. Conjunctival staining revealed minimal ocular surface irregularities. Her postoperative care otherwise was completely uneventful, and she continued using PF artificial tears and Restasis®. At 1 year of postoperative care, the patient did not require additional medications. However, she continues to use her existing medications to manage her residual dry eye symptoms.
Patient #2
A 25-year-old Caucasian male contact lens wearer, with a CFTR-RD diagnosis per patient since childhood, presented seeking refractive surgery. He did not complain of any ocular or systemic issues of CFTR-RD. The patient stated he was not taking CF medications and had no family history of CF. Preoperatively, his manifest refraction was −2.75 D OU and his corneal thickness was approximately 585 μm OU. Corneal and conjunctival staining revealed no remarkable findings. His Schirmer’s test was above 10 mm in 5 minutes and his TBUT was greater than 7 seconds. LASIK was performed using an iFS® Advanced Femtosecond IntraLase™ Laser (Johnson & Johnson Surgical Vision Inc., Santa Ana, CA) to create an 8.7 mm diameter flap with a flap thickness of 110 μm. A VISX STAR S3™ (Johnson & Johnson Surgical Vision Inc., Santa Ana, CA) was used to create a 6.0 mm ablation zone with a transitional zone of 8.0 mm.
Postoperatively the patient responded well but developed more dry eye symptoms relative to other LASIK patients, prompting the insertion of punctal plugs superiorly and inferiorly for approximately 6–7 months. Afterward, Refresh Plus PF Lubricant Eye Drops were prescribed for residual dryness. He had no corneal or conjunctival staining. One year postoperatively, the patient presented with 20/20 corrected vision and a refractive error of +0.5 D OD and +0.25 D OS.
Discussion
We acknowledge that while patients #1 and #2 had successful surgical outcomes and appropriate treatment, it is important to note that both had mild clinical manifestations of CF and CFTR-RD, respectively. The surgeon’s approach at that time was untested due to a lack of guidelines and general knowledge concerning CF/CFTR-RD and their potential multisystem pathophysiology, particularly their effects on the cornea and eye. To our best knowledge, there is no literature discussing refractive surgery considerations in patients with CF/CFTR-RD. We propose a structured approach to assessing and treating this patient population in preparation for refractive surgery (Figure 1). This approach can be applied to mild to severe CF cases, as well as CFTR-RD, and may assist clinicians who encounter this patient population. We also propose a simple novel surgical risk scoring system that may be used in-office with these patients (Table 1).
Ophthalmologists considering refractive surgery for these patients should be familiar with the non-ocular systemic manifestations of CF. These may include pulmonary pathologies such as chronic obstructive pulmonary disease (COPD), bronchiectasis, or recurrent pulmonary infections. Other manifestations include hepatic disease, pancreatic insufficiency, CF-related diabetes, malnutrition, and vitamin A malabsorption and deficiency.3,8,15 In contrast, patients with CFTR-RD generally have single organ system involvement and milder symptoms than those with CF. Typical manifestations include mild pulmonary issues, congenital bilateral absence of the vas deferens, disseminated bronchiectasis, nasal polyps, chronic sinusitis, and acute recurrent or chronic pancreatitis.7,13 Patient #1 and patient #2 both exemplify how these systemic manifestations may be minimal or absent in well-controlled or less severe CF/CFTR-RD cases. Screening for these manifestations during a medical history and coordinating care with these patients’ primary care physicians, pulmonologists, medical geneticists, or other medical professionals will help determine their refractive surgery eligibility. ROS questions for screening CF/CFTR-RD patients are listed in Table 2. Lastly, genetic testing, if not already performed, may also help guide systemic treatment based on specific CFTR variants.16 Although research has shown that certain CFTR gene variants correlate with more severe gastrointestinal manifestations rather than pulmonary ones, further research is indicated to determine which genotypes may correlate with more severe ocular pathology.9
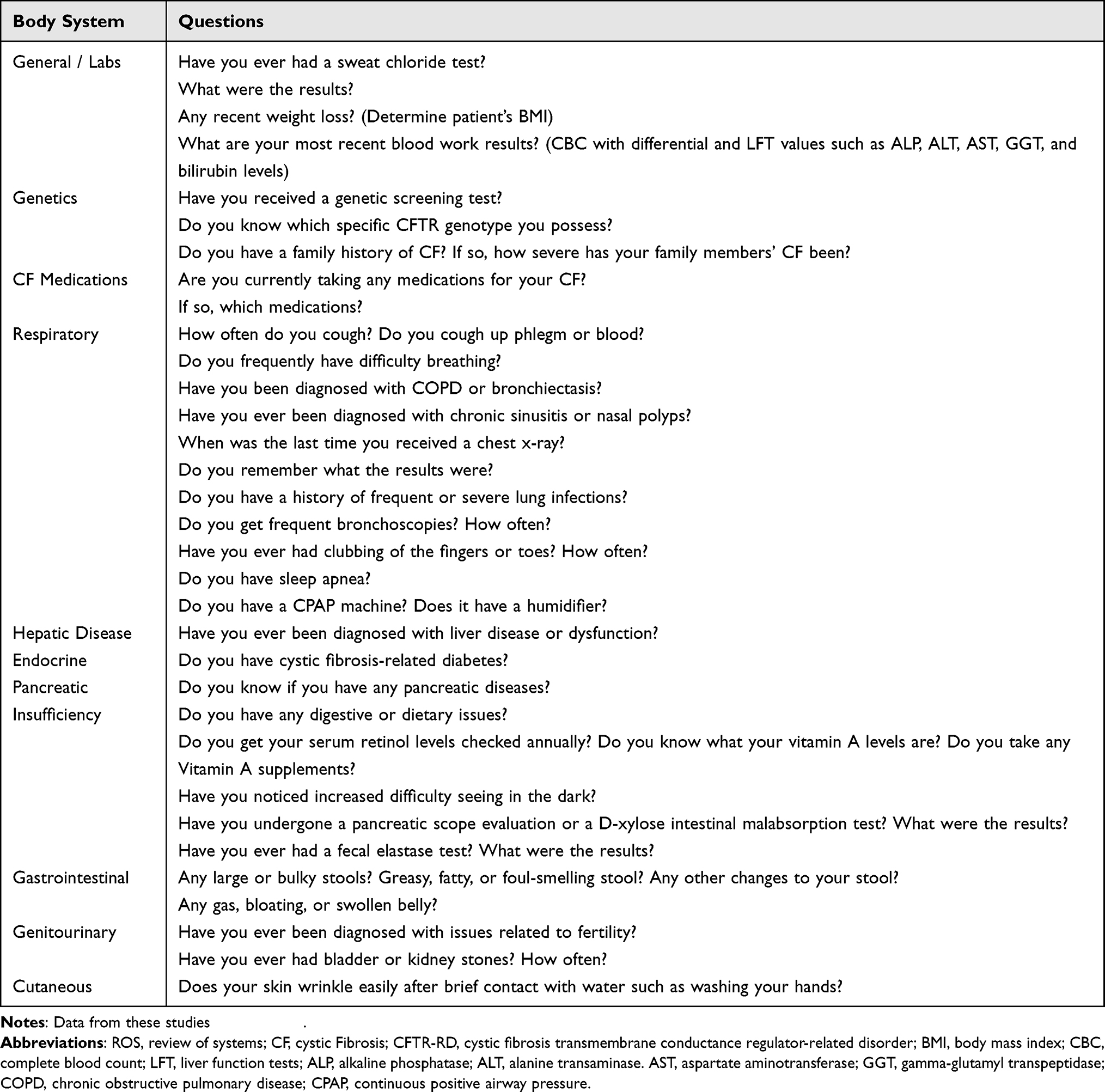 |
Table 2 ROS Questions to Consider When Taking a CF/CFTR-RD Patient’s History |
Hypoxemia and malnutrition, potential systemic manifestations of CF/CFTR-RD, underlie some ocular pathologies. Patients with more severe CF/CFTR-RD phenotypes may experience corneal hypoxia during sleep. Corneal hypoxia can lead to stromal acidosis and stromal thinning.17 A study by Bradley et al found that patients with moderately severe CF can experience hypercapnia and hypoxemia during sleep and exercise.18 Another study showed that obstructive sleep apnea (OSA), a condition for which patients with CF may be predisposed, is significantly correlated with peripapillary retinal nerve fiber layer thinning, increased risk of acute retinal vein occlusion, decreased endothelial cell density, and decreased central corneal thickness due to hypoxia.19–21 Corneal damage may be prevented by ensuring that patients with moderate to severe CF/CFTR-RD are screened for OSA and receive adequate treatment. As CF patients are predisposed to developing OSA, screening them for floppy eyelid syndrome (FES) and keratoconus (KCN) may be indicated as these conditions have been significantly correlated with OSA.21,22 Proposed mechanisms for FES and KCN suggest that inflammatory mediators, such as matrix metalloproteinases (MMPs), may play a role in their pathogenesis, which can be elevated in CF patients.22,23 Regarding malnutrition, vitamin A deficiency can cause xerophthalmia, conjunctival xerosis, and night blindness.15,24 These can be screened for while performing a medical history and physical exam. In-office tests that can screen for possible vitamin A deficiency include dark adaptation and contrast sensitivity. Patients with vitamin A deficiency secondary to CF may have abnormal dark adaptation and reduced contrast sensitivity.25,26 A vitamin A blood test can also be utilized.
CFTR dysfunction can also cause ocular pathologies. CFTR expression and activity have been observed in lacrimal and meibomian glands, and other secretory epithelial cells in the eye.15,20,27,28 The hyperviscous mucus secretions associated with dysfunctional CFTR proteins may obstruct these glands and cells, leading to lacrimal acinar cell degeneration, meibomian gland dysfunction (MGD), and decreased glandular secretions. Reducing lipid, aqueous, and mucosal glandular secretions destabilizes the tear-film, decreases ocular surface nutrient delivery, and decreases ocular surface lubrication, respectively.15 These obstructions may also trigger a pro-inflammatory response, leading to decreased corneal epithelial cell integrity, conjunctival goblet cell loss, and blepharitis.15,20,28
These alterations can cause ocular surface disorders (OSDs), such as concurrent aqueous-deficient DED and evaporative DED, also known as mixed etiology DED, and superficial punctate keratitis, which was found in patient #1.15,20,29 Additionally, patient #1 was noted to have FK before and after PRK surgery. It has been proposed that an increased tear film mucus-to-aqueous ratio precedes FK. This altered ratio can cause a relative increase in tear film mucus viscosity and impaired mucus clearance, thereby damaging epithelial cells and precipitating filament growth in the cornea.30 FK onset is most commonly associated with aqueous-deficient DED, suggesting that patients with CF/CFTR-RD are at a higher risk of developing FK.31 The corneal surface damage sustained during refractive surgery may exacerbate pre-existing OSDs in these patients, which could be a contraindication for surgery.32–34 A meta-analysis on dry eye occurrence after refractive surgery determined that LASIK had significant reductions in both TBUT and tear production while PRK did not. There has also been an increased rate of dry eye in LASIK compared to other refractive surgery methods, including PRK.35 While this meta-analysis indicates that PRK may be preferred over LASIK in CF and CFTR-RD patients due to the decreased risk of post-operative dry eye complications, the procedure of choice is at the discretion of the surgeon on a case-by-case basis.
A structured approach to assessing and managing this patient population in preparation for refractive surgery is presented in Figure 1. Patients with CF/CFTR-RD who are seeking refractive surgery should be screened with a Standard Patient Evaluation of Eye Dryness (SPEED) II or Ocular Surface Disease Index (OSDI) questionnaire to assess for dry eye symptoms. Tear osmolarity tests, Schirmer’s tests, vital dye stains, TBUT measurements, and meibography can be instrumental in revealing ocular pathophysiology.20,36,37 In both patient cases, initial vital dye stains played a key role in the clinical decision-making progress, prompting further treatment for patient #1 and an indication for safe surgical intervention for patient #2. Postoperative dry eye screening in patient #2 also revealed dry eye symptoms which were successfully managed. Lastly, impression cytology can screen for abnormal changes in conjunctival goblet cell number and density.38,39
Markers of ocular surface inflammation should also be assessed, as some are elevated in patients with CF. InflammaDry® (Quidel Corp., San Diego, CA) kits can be used to screen for matrix metalloproteinase 9 (MMP-9) which is elevated in ocular surface inflammation and MGD.23,37 Several studies have also shown that patients with CF have elevated levels of inflammatory mediators interleukin-8 (IL-8), interferon-γ (IFN-γ), macrophage inflammatory protein-1α (MIP-1α), and MIP-1β in their tear films. These may be measured using ELISA assays to further assess the severity of each patient’s ocular surface inflammation.40–42 While ELISA assays are not readily available for clinical use, they may play instrumental roles in diagnosing OSD in the future. Table 3 summarizes relevant screening tools for evaluating patients with CF/CFTR-RD and the possible positive findings.
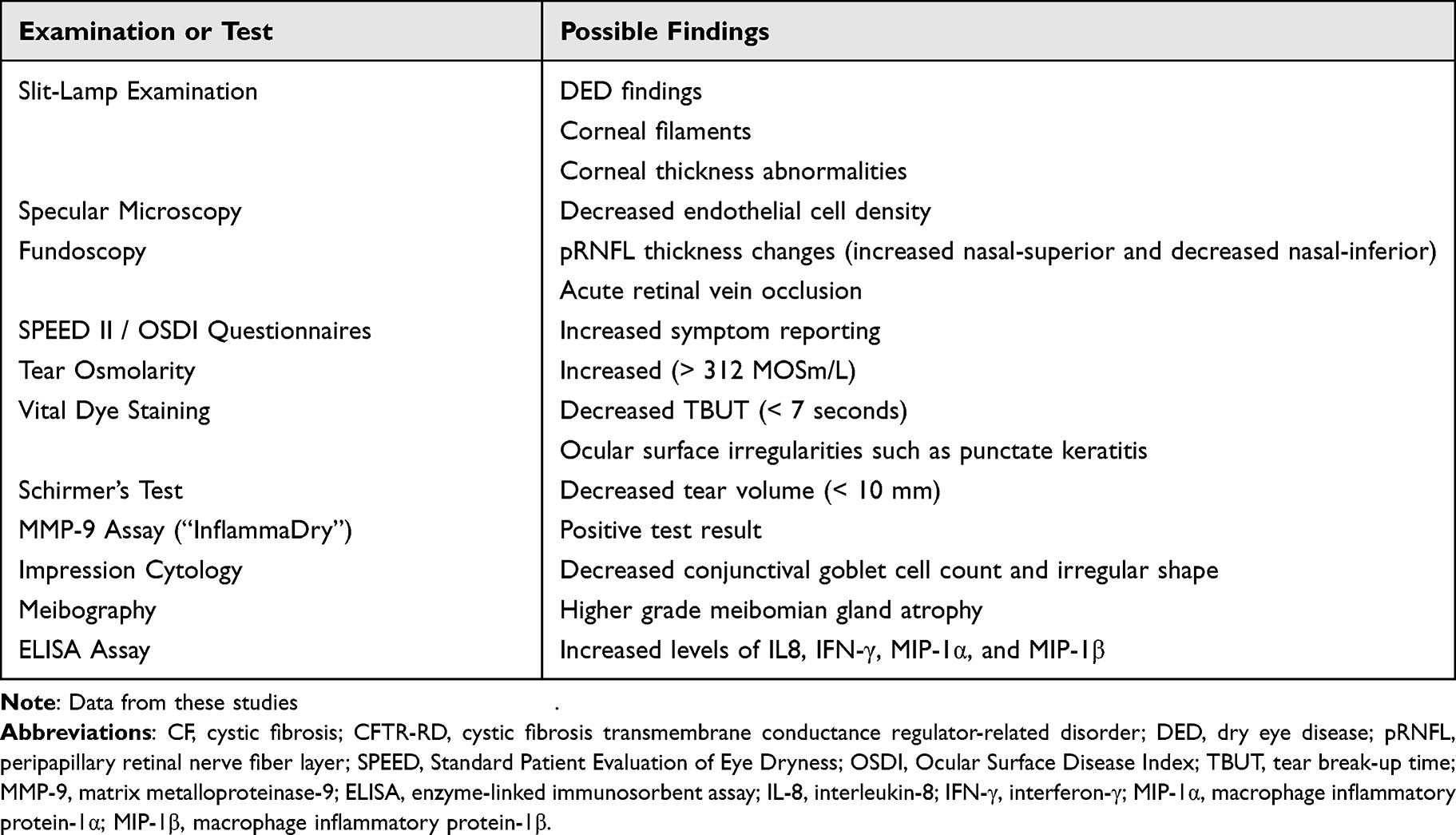 |
Table 3 Overview of Ophthalmologic Screening Tools for CF/CFTR-RD Patients |
A new potential tool for screening patients with CF/CFTR-RD for refractive surgery is a novel surgical risk scoring system (Table 1). This simple, novel scoring system was adapted from the Shwachman-Kulczycki (SK) score, which was created in 1958 to assess the clinical severity of CF in children.43 Although the SK score was created over 60 years ago, it has remained a useful and simple tool for monitoring the clinical severity of CF in children and adolescents.44 Our modified scoring system is comprised of three categories: general activity, physical examination, and nutrition. The score from each section is summed to obtain a final score, after which the surgical risk of the patient is determined to be low (3), moderate (4–6), or high (7–9). Our scoring system may assist in refractive surgery risk stratification because inflammatory mediators in tears, such as IL-8, IFN-γ, and MIP-1α, have been shown to be elevated with increasing clinical severity and dry eye findings in patients with CF who were assessed using a similar scoring system.41,45 Our scoring system has not yet been validated and may serve as a starting point for others to refine in the future.
Following screening, various treatment strategies may be employed to prepare them for surgery and ensure a rapid and uneventful recovery. While nebulized N-acetylcysteine products, such as Mucomyst® (E.R. Squibb & Sons, LLC, New Brunswick, NJ), are indicated for controlling pulmonary mucus obstructions, topical N-acetylcysteine medications, which were utilized in patient #1, can also be used to manage inflammatory CF/CFTR-RD ocular manifestations by inhibiting MMP-9 and nuclear factor kappa B (NF-κB).23,46,47 Doxycycline also inhibits MMP-9, providing a similar protective effect for the cornea, and may be used preoperatively to treat bacterial blepharitis.37,48,49 Mainstream treatments for various ocular surface disorders should be implemented as first-line treatment in these patients, including artificial tears, topical steroids, cyclosporin A 0.05% (Restasis®), lifitegrast 5% (Novartis Pharmaceuticals Corporation, Xiidra ®, East Hanover, NJ)), punctal plugs, and warm compresses.37
Unlike patients #1 and #2, some may experience unexpectedly severe or recalcitrant perioperative OSDs. Additional treatment options are available in these circumstances, including autologous blood serum drops, platelet-rich plasma (PRP) eye drops, therapeutic contact lens bandages, self-retaining amniotic membranes, amniotic membrane extract eye drops (AMEED), and topical mesenchymal stem cell therapies, such as the commercial product Regener-Eyes® (Regenerative Processing Plant, LLC, Palm Harbor, FL).50–56 AMEED and Regener-Eyes® are not currently FDA approved but have promising therapeutic potential as they leverage their anti-inflammatory, anti-angiogenic, and non-immunogenic properties to promote limbal stem cell proliferation, facilitate corneal epithelial healing, and treat DED.54–56
Further research may identify safer and more effective drugs for treating CF’s ocular manifestations by targeting the CFTR protein, as ivacaftor, for example, has been correlated with cataract development in children and adolescents.57 Multiple novel CFTR-targeting ocular treatments are also undergoing research and may be utilized to treat CF/CFTR-RD in the future. Small molecule CFTR activators treat DED by increasing ocular surface membrane hydration but require further research in patients with CF/CFTR-RD.58
To our knowledge, the two patient cases detailing refractive surgery considerations and outcomes in individuals with CF and CFTR-RD are the first to be reported. Successful completion of PRK and LASIK in these patients offers encouragement that refractive surgery can be completed in other patients with CF/CFTR-RD. However, the lack of knowledge and precedent regarding this topic makes it difficult for ophthalmologists to validate whether patients with CF/CFTR-RD are eligible for these procedures. Therefore, this article proposes a novel structured approach for assessing and treating patients with CF/CFTR-RD in preparation for refractive surgery, based on the most current literature (Figure 1). Further research will hopefully lead to the creation of systematic guidelines to provide the best possible outcomes for these patients. Finally, given the relatively unknown association between CF/CFTR-RD and refractive surgery outcomes, we suggest that ophthalmologists and patients with CF/CFTR-RD discuss the risks and unknown complications of these procedures in detail before proceeding with surgery. We also encourage surgeons who perform refractive surgery on these patients to consider publishing reports on their outcomes to help establish precedent on this topic.
Data Sharing Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Compliance with Ethics Guidelines
The case series was approved by the Hoopes Vision Ethics Board and patients’ consent to publish was acquired.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This article is supported by Research to Prevent Blindness, New York, USA.
Disclosure
All authors declare that they have no conflict of interest in this work.
References
1. Scotet V, L’hostis C, Férec C. The changing epidemiology of cystic fibrosis: incidence, survival and impact of the CFTR gene discovery. Genes. 2020;11(6):6. doi:10.3390/GENES11060589
2. MacKenzie T, Gifford AH, Sabadosa KA, et al. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry. Ann Intern Med. 2014;161(4):233–241. doi:10.7326/M13-0636
3. Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet. 2021;397(10290):2195–2211. doi:10.1016/S0140-6736(20)32542-3
4. Levin MH, Verkman AS. Aquaporins and CFTR in ocular epithelial fluid transport. J Membr Biol. 2006;210(2):105–115. doi:10.1007/S00232-005-0849-1/TABLES/1
5. Veit G, Avramescu RG, Chiang AN, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424. doi:10.1091/MBC.E14-04-0935
6. Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18(2):81. doi:10.1016/J.MOLMED.2011.10.003
7. Bombieri C, Claustres M, de Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10(SUPPL. 2):S86–S102. doi:10.1016/S1569-1993(11)60014-3
8. Brennan M-L, Schrijver I. Cystic fibrosis: a review of associated phenotypes, use of molecular diagnostic approaches, genetic characteristics, progress, and dilemmas. J Mol Diagn. 2016;18(1):3–14. doi:10.1016/J.JMOLDX.2015.06.010
9. Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. 2010;1214(1):57. doi:10.1111/J.1749-6632.2010.05879.X
10. Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the cystic fibrosis foundation. J Pediatr. 2017;181:S4–S15.e1. doi:10.1016/J.JPEDS.2016.09.064
11. Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis foundation consensus report. J Pediatr. 2008;153(2):S4. doi:10.1016/J.JPEDS.2008.05.005
12. Levy H, Nugent M, Schneck K, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet. 2016;89(5):539. doi:10.1111/CGE.12711
13. Kilinc AA, Alishbayli G, Taner HE, Cokugras FC, Cokugras H. Clinical characteristics and genetic analysis of cystic fibrosis transmembrane conductance reseptor-related disease. Pediatr Int. 2020;62(5):629–633. doi:10.1111/PED.14173
14. Marshall B, Faro A, Brown W, et al. Cystic fibrosis foundation patient registry 2020 annual data report; 2020. Available from: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf.
15. Alghadyan A, Aljindan M, Alhumeidan A, Kazi G, Mcmhon R. Lacrimal glands in cystic fibrosis. Saudi J Ophthalmol. 2013;27:113–116. doi:10.1016/j.sjopt.2013.01.001
16. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2020;10:1662. doi:10.3389/FPHAR.2019.01662/BIBTEX
17. Efron N, Ang JHB. Corneal hypoxia and hypercapnia during contact lens wear. Optom Vis Sci. 1990;67(7):512–521. doi:10.1097/00006324-199007000-00009
18. Bradley S, Solin P, Wilson J, Johns D, Walters EH, Naughton MT. Hypoxemia and hypercapnia during exercise and sleep in patients with cystic fibrosis. Chest. 1999;116(3):647–654. doi:10.1378/CHEST.116.3.647
19. Bojarun A, Vieversyte Z, Jaruseviciene R, Galgauskas S, Asoklis R, Zablockis R. Effect of obstructive sleep apnea on corneal morphological characteristics. Cornea. 2019;38(12):1576–1581. doi:10.1097/ICO.0000000000002069
20. Giannakouras P, Kanakis M, Diamantea F, et al. Ophthalmologic manifestations of adult patients with cystic fibrosis. Eur J Ophthalmol. 2021;32(2):976–983. doi:10.1177/11206721211008780
21. Shakkottai A, Irani S, Nasr SZ, O’Brien LM, Chervin RD. Risk factors for obstructive sleep apnea in cystic fibrosis. Pediatr Pulmonol. 2022;57(4):926–934. doi:10.1002/PPUL.25811
22. Liu PK, Chiu TY, Wang NK, Levi SR, Tsai MJ. Ocular complications of obstructive sleep apnea. J Clin Med. 2021;10:15. doi:10.3390/JCM10153422
23. Ramaesh T, Ramaesh K, Riley SC, West JD, Dhillon B. Effects of N-acetylcysteine on matrix metalloproteinase-9 secretion and cell migration of human corneal epithelial cells. Eye. 2012;26(8):1138–1144. doi:10.1038/EYE.2012.135
24. Wamsley S, Patel SM, Wood MG, Villalobos R, Albert DM, Mootha VV. Advanced keratomalacia with descemetocele in an infant with cystic fibrosis; 2005. Available from: https://jamanetwork.com/.
25. Huet F, Semama D, Maingueneau C, Charavel A, Nivelon JL. Vitamin A deficiency and nocturnal vision in teenagers with cystic fibrosis. Eur J Pediatr. 1997;156(12):949–951. doi:10.1007/S004310050749
26. Leguire LE, Pappa KS, Kachmer ML, Rogers GL, Bremer DL. Loss of contrast sensitivity in cystic fibrosis. Am J Ophthalmol. 1991;111(4):427–429. doi:10.1016/S0002-9394(14)72375-X
27. Turner HC, Bernstein A, Candia OA. Presence of CFTR in the conjunctival epithelium. Curr Eye Res. 2002;24(3):182–187. doi:10.1076/CEYR.24.3.182.8297
28. Mrugacz M, Kasacka I, Bakunowicz-Lazarczyk A, Kaczmarski M, Kulak W. Impression cytology of the conjunctival epithelial cells in patients with cystic fibrosis. Eye. 2008;22:1137–1140. doi:10.1038/sj.eye.6702867
29. Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Dtsch Arztebl Int. 2015;112(5):71. doi:10.3238/ARZTEBL.2015.0071
30. Weiss M, Molina R, Ofoegbuna C, Johnson DA, Kheirkhah A. A review of filamentary keratitis. Surv Ophthalmol. 2022;67(1):52–59. doi:10.1016/J.SURVOPHTHAL.2021.04.002
31. Albietz J, Sanfilippo P, Troutbeck R, Lenton LM. Management of filamentary keratitis associated with aqueous-deficient dry eye. Optom Vis Sci. 2003;80(6):420–430. doi:10.1097/00006324-200306000-00007
32. Garcia-Zalisnak D, Nash D, Yeu E. Ocular surface diseases and corneal refractive surgery. Curr Opin Ophthalmol. 2014;25(4):264–269. doi:10.1097/ICU.0000000000000077
33. Murakami Y, Manche EE. Prospective, randomized comparison of self-reported postoperative dry eye and visual fluctuation in LASIK and photorefractive keratectomy. Ophthalmology. 2012;119(11):2220–2224. doi:10.1016/J.OPHTHA.2012.06.013
34. Shah R. History and results; indications and contraindications of SMILE compared with LASIK. Asia Pac J Ophthalmol. 2019;8(5):371. doi:10.1097/01.APO.0000580132.98159.FA
35. Sambhi RDS, Sambhi GDS, Mather R, Malvankar-Mehta MS. Dry eye after refractive surgery: a meta-analysis. Can J Ophthalmol. 2020;55(2):99–106. doi:10.1016/J.JCJO.2019.07.005
36. Vunnava KP, Shetty N, Kapur KB. A review of meibography for a refractive surgeon. Indian J Ophthalmol. 2020;68(12):2663. doi:10.4103/IJO.IJO_2465_20
37. Starr CE, Gupta PK, Farid M, et al. An algorithm for the preoperative diagnosis and treatment of ocular surface disorders. J Cataract Refract Surg. 2019;45(5):669–684. doi:10.1016/J.JCRS.2019.03.023
38. Nelson JD, Wright JC. Conjunctival goblet cell densities in ocular surface disease. Arch Ophthalmol. 1984;102(7):1049–1051. doi:10.1001/ARCHOPHT.1984.01040030851031
39. Doughty MJ. Assessment of goblet cell size and density in relation to epithelial cell (multi) layering on conjunctival impression cytology samples. Graefes Arch Clin Exp Ophthalmol. 2020;258(8):1727–1734. doi:10.1007/S00417-020-04725-5
40. Hagan S, Martin E, Enríquez-de-Salamanca A. Tear fluid biomarkers in ocular and systemic disease: potential use for predictive, preventive and personalised medicine. EPMA J. 2016;7(1). doi:10.1186/S13167-016-0065-3
41. Mrugacz M, Zelazowska B, Bakunowicz-Lazarczyk A, Kaczmarski M, Wysocka J. Elevated tear fluid levels of MIP-1alpha in patients with cystic fibrosis. J Interferon Cytokine Res. 2007;27(6):491–495. doi:10.1089/JIR.2007.0149
42. Mrugacz M. CCL4/MIP-1beta levels in tear fluid and serum of patients with cystic fibrosis. J Interferon Cytokine Res. 2010;30(7):509–512. doi:10.1089/JIR.2009.0102
43. Shwachman H, Kulczycki LL. Long-term study of one hundred five patients with cystic fibrosis: studies made over a five- to fourteen-year period. AMA J Dis Child. 1958;96(1):6–15. doi:10.1001/ARCHPEDI.1958.02060060008002
44. Stollar F, Adde FV, Cunha MT, Leone C, Rodrigues JC. Shwachman-Kulczycki score still useful to monitor cystic fibrosis severity. Clinics. 2011;66(6):979–983. doi:10.1590/S1807-59322011000600010
45. Mrugacz M, Kaczmarski M, Bakunowicz-Lazarczyk A, Zelazowska B, Wysocka J, Minarowska A. IL-8 and IFN-gamma in tear fluid of patients with cystic fibrosis. J Interferon Cytokine Res. 2006;26(2):71–75. doi:10.1089/JIR.2006.26.71
46. Crist AJ. Case report: management of refractory filamentary keratitis with N-acetylcysteine. Optom Vis Sci. 2021;98(6):547–551. doi:10.1097/OPX.0000000000001701
47. Eghtedari Y, Oh LJ, Girolamo N, Watson SL. The role of topical N-acetylcysteine in ocular therapeutics. Surv Ophthalmol. 2022;67(2):608–622. doi:10.1016/J.SURVOPHTHAL.2021.07.008
48. de Paiva CS, Corrales RM, Villarreal AL, et al. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp Eye Res. 2006;83(3):526–535. doi:10.1016/J.EXER.2006.02.004
49. Beringer PM, Owens H, Nguyen A, Benitez D, Rao A, D’Argenio DZ. Pharmacokinetics of doxycycline in adults with cystic fibrosis. Antimicrob Agents Chemother. 2012;56(1):70. doi:10.1128/AAC.05710-11
50. Noble BA, Loh RSK, MacLennan S, et al. Comparison of autologous serum eye drops with conventional therapy in a randomised controlled crossover trial for ocular surface disease. Br J Ophthalmol. 2004;88(5):647–652. doi:10.1136/BJO.2003.026211
51. Alio L, Arnalich-Montiel F E, Rodriguez A. The role of “eye platelet rich plasma” (E-PRP) for wound healing in ophthalmology. Curr Pharm Biotechnol. 2012;13(7):1257–1265. doi:10.2174/138920112800624355
52. Kim JS, Kim JC, Na BK, Jeong JM, Song CY. Amniotic membrane patching promotes healing and inhibits proteinase activity on wound healing following acute corneal alkali burn. Exp Eye Res. 2000;70(3):329–337. doi:10.1006/EXER.1999.0794
53. Arora R, Jain S, Monga S, Narayanan R, Raina UK, Mehta DK. Efficacy of continuous wear PureVision contact lenses for therapeutic use. Cont Lens Anterior Eye. 2004;27(1):39–43. doi:10.1016/J.CLAE.2003.09.004
54. Sabater-Cruz N, Figueras-Roca M, Ferrán-Fuertes M, et al. Amniotic membrane extract eye drops for ocular surface diseases: use and clinical outcome in real-world practice. Int Ophthalmol. 2021;41(9):2973–2979. doi:10.1007/S10792-021-01856-4/TABLES/3
55. Asl NS, Nejat F, Mohammadi P, et al. Amniotic membrane extract eye drop promotes limbal stem cell proliferation and corneal epithelium healing. Cell J. 2019;20(4):459–468. doi:10.22074/CELLJ.2019.5423
56. Harrell CR, Jankovic MG, Fellabaum C, et al. Molecular mechanisms responsible for anti-inflammatory and immunosuppressive effects of mesenchymal stem cell-derived factors. Adv Exp Med Biol. 2019;1084:187–206. doi:10.1007/5584_2018_306/COVER/
57. Guevera MT, Mccolley SA. The safety of lumacaftor and ivacaftor for the treatment of cystic fibrosis HHS Public Access. Expert Opin Drug Saf. 2017;16(11):1305–1311. doi:10.1080/14740338.2017.1372419
58. Flores AM, Casey SD, Felix CM, Phuan PW, Verkman AS, Levin MH. Small-molecule CFTR activators increase tear secretion and prevent experimental dry eye disease. FASEB J. 2016;30(5):1789. doi:10.1096/FJ.201500180
59. Daye M, Pekcan S, Mevlitoğlu İ. Skin findings in cystic fibrosis cases. J Geophys Res. 2018;123(5):1012–1040. doi:10.5152/cjms.2018.596
60. Lipnick RN, Glass RBJ. Bone changes associated with cystic fibrosis. Skeletal Radiol. 1992;21(2):115–116. doi:10.1007/BF00241837
61. Starr MR, Norby SM, Scott JP, Bakri SJ. Acute retinal vein occlusion and cystic fibrosis. Int J Retina Vitreous. 2018;4:26. doi:10.1186/s40942-018-0129-8
62. Bron AJ, Abelson MB, Ousler G, et al. Methodologies to diagnose and monitor dry eye disease: report of the diagnostic methodology subcommittee of the international Dry Eye Workshop (2007). Ocular Surface. 2007;5(2):108–152. doi:10.1016/S1542-0124(12)70083-6
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.