")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 16
The Integral Role of Chloride & With-No-Lysine Kinases in Cell Volume Regulation & Hypertension
Authors Koulouridis I , Koulouridis E
Received 18 April 2023
Accepted for publication 28 July 2023
Published 14 August 2023 Volume 2023:16 Pages 183—196
DOI https://doi.org/10.2147/IJNRD.S417766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Ioannis Koulouridis, Efstathios Koulouridis
Private Nephrology Office, Corfu, Greece
Correspondence: Ioannis Koulouridis, Spirou Rath 41, Corfu, 49100, Greece, Tel +30 6944276642 ; +30 2661022660, Email [email protected]; [email protected]
Abstract: Chloride anions are the most abundant in humans. For many years, it has been believed that chloride is simply a counterion of all other cations, ensuring the electroneutrality of the extracellular space. Recent data suggests that chloride anions possess a broad spectrum of important activities that regulate vital cellular functions. It is now evident that, apart from its contribution to the electroneutrality of the extracellular space, it acts as an osmole and contributes to extracellular and intracellular volume regulation. Its anionic charge also contributes to the generation of cell membrane potential. The most interesting action of chloride anions is their ability to regulate the activity of with-no-lysine kinases, which in turn regulate the activity of sodium chloride and potassium chloride cotransporters and govern the reabsorption of salt and excretion of potassium by nephron epithelia. Chloride anions seem to play a crucial role in cell functions, such as cell volume regulation, sodium reabsorption in the distal nephron, potassium balance, and sodium sensitivity, which lead to hypertension. All of these functions are accomplished on a molecular level via complicated metabolic pathways, many of which remain poorly defined. We attempted to elucidate some of these pathways in light of recent advances in our knowledge, obtained mainly from experimental studies.
Keywords: intracellular chloride, cell volume regulation, WNK kinases, extracellular potassium, salt sensitive hypertension
Introduction
The regulation of intracellular volume is crucial for cell survival. Cells are extremely sensitive to subtle changes in osmotic pressure, which affect their shape and function. Early studies have shown that the intracellular volume is effectively regulated by the movement of potassium and chloride ions through the cell membrane and via the de novo synthesis of certain osmolytes. Quick movement of osmotically active ions results in fast restoration of osmolal changes in the intracellular space.1
Potassium cations are primarily distributed in the intracellular space, whereas chloride anions are abundant in the extracellular space. This discrepancy makes chloride and potassium ions the first aid for rescue in circumstances of acute intracellular volume disturbances. Chloride transport through the cell membrane is accomplished via specialized channels, antiporters, and sodium chloride co-transporters. At the level of kidney epithelia, the main carriers of intracellular chloride movement are cation-chloride cotransporters (CCCs), which in turn are regulated by the activation of with-no-lysine kinases (WNKs).2–4
Nephron epithelial cells must reabsorb large amounts of electrolytes, organic molecules, and water to achieve an effective balance between sodium and water, which is a prerequisite for survival of mammalian species in terrestrial life. Furthermore, the discrepancy in potassium distribution between intracellular and extracellular spaces requires strict regulation of potassium levels in the extracellular space because of harmful consequences in cases of concentration derangements. Although the reabsorption of these substances is achieved via cellular and paracellular pathways, the main reabsorption of sodium, potassium, and chloride in the thick ascending limb of Henle’s loop (TAL) and the fine-tuning of sodium reabsorption and potassium excretion in the distal nephron is achieved via the cellular pathway. In specific, according to the passive model of urine concentration, the ascending limb of Henle’s loop is impermeable to water, and the active transport of sodium, potassium, and chloride ions by the NKCC2 cotransporter gradually dilutes the tubular fluid. As a result, the tubular fluid reaching the distal convoluted tubule (DCT) becomes hypotonic compared to the plasma. The DCT epithelial cells are exposed to a continuous osmotic challenge due to the impermeability of the ascending limb to water and the activity of the sodium-chloride cotransporter (NCC), which increases the burden of osmotically active ions (Na and Cl). This means that these cells are exposed to a tremendous osmotic challenge during the day, and first, they must rescue themselves from cell volume derangements.5,6
It is worth noting that the trafficking of osmoles through the cell body is not automatic and requires the activation of specific transporters and ion channels, regulated by metabolic pathways triggered by signaling mechanisms. De Smet et al demonstrated in 1995 that kidney cells can rapidly respond to changes in volume under hypotonic conditions, indicating their ability to regulate volume in response to osmotic challenges.7 Recent experimental findings by Lopez-Cayuqueo et al revealed the expression of LRRC8/VRAC channels in rat proximal tubules and distal nephron intercalated cells, and their deletion resulted in proximal tubular injury, resembling a mild Fanconi-like syndrome.8 These studies provide valuable insights into the volume regulation mechanisms of kidney epithelial cells and their implications in renal physiology and pathology.
In cases of increased potassium consumption, the kidneys play a crucial role in maintaining extracellular volume and potassium homeostasis to achieve a delicate balance between the conservation of extracellular volume and the excretion of excess potassium. According to the literature mentioned in the paragraph above as well as newer data presented by Baturina GS et al9 kidney epithelial cells exhibit the ability of cell volume regulation from the proximal tubule to the distal tubule and collecting duct, although the osmotic challenge is not equal for each individual segment of the nephron. This variability implies that different nephron epithelial cells may prioritize tasks differently. However, it is a logical assumption that every cell prioritizes first its survival and then the teleological purpose dedicated by nature. Therefore, in cases of increased potassium consumption, it is likely that nephron epithelial cells prioritize their tasks according to the following scheme: cell volume maintenance > potassium homeostasis > extracellular volume maintenance. This assumption is also supported by the aldosterone paradox and the natriuretic effect of high potassium ingestion, which indicate that distal nephron epithelial cells prioritize extracellular potassium homeostasis over extracellular volume homeostasis, even in settings of increased salt consumption. However, we acknowledge that in certain extreme situations, such as certain types of shock, where cell volume maintenance becomes a higher priority, the administration of vasopressors can enhance NaCl reabsorption in the proximal tubules and thick ascending limbs. This leads to decreased Na delivery to distal tubules and impairment of K secretion. These examples highlight the complex interplay of various factors influencing task prioritization in nephron epithelial cells under different physiological conditions.
In this review article, we attempted to elucidate the steps pointing to the importance of chloride anions, which accompany sodium and mediate its capacity to increase blood pressure under conditions of excess salt consumption. We review the discoveries that emphasize the importance of chloride anions in cell volume regulation and potassium homeostasis in the distal nephron. By synthesizing the available literature, we provide a comprehensive summary of how chloride anion and the intake of potassium interplay with one another, and how these ions collectively affect sodium sensitivity. Understanding this interplay has significant implications associated with the dietary practices of modern humans.
Cell Volume Regulation and Chloride Anion
The maintenance of cellular volume is fundamental for proper cell function and survival. The lipid bilayer of the cell membrane separates the cell from the environment and behaves as a semipermeable membrane, meaning that it is freely permeable to water and impermeable to inorganic and organic solutes. Because the cell membrane is unable to produce significant hydrostatic pressure, any disturbance in solute concentration across each side results in an osmotic gradient with consequent movement of water towards the more concentrated compartment to achieve a new osmotic equilibrium. During this process, the cells swell or shrink according to the gain or loss of water from the intracellular space.1
Cell volume disturbances are grouped into two distinct categories based on osmotic pressure perturbations: isosmotic and anisosmotic. Isosmotic disturbances are a consequence of altered intracellular osmolyte content, whereas anisosmotic cell volume disturbances result from extracellular osmotic dysregulation.1
Cells are very sensitive to volume alterations and are capable of sensing and responding quickly, even after very small changes, as low as 1–2% of the normal volume size.4 Changes in cell volume recruit specific mechanisms for volume restoration known as Regulatory Volume Decrease (RVD) and Regulatory Volume Increase (RVI), depending on the status of the cell. RVD is triggered after cell swelling as a consequence of hypotonic stress, whereas RVI is triggered after cell shrinkage as a consequence of hypertonic stress.2
Volume repair mechanisms encompass the specific movement of solutes in two categories: inorganic ions and organic osmolytes such as taurine and glycine. The movement of inorganic ions through the cell membrane was rapid, within seconds. Organic osmolytes require a longer time, usually many hours, after initial activation.2 This is because specific transporters of inorganic ions are present either in the cell membrane or in the cell cytoplasm and are readily recruited for ionic transport, whereas organic osmolytes require the activation of specific genes and enzymes for their synthesis. While it is true that major electrolytes, including inorganic ions, are essential for cellular function, their intracellular concentrations must be tightly regulated. Accumulation of a large amount of inorganic ions within the cell can disrupt the electric charge of the cell membrane, intracellular pH, and protein function. In this context, excessive accumulation of inorganic ions can be harmful to the cell. Organic osmolytes, on the other hand, are compatible with normal cell function even at high concentrations and do not pose the same risks.1,2 Thus, it is crucial for cells to maintain precise control over intracellular ion concentrations to ensure proper cellular homeostasis and function, rather than relying on excessive accumulation of inorganic ions to counteract severe or prolonged volume derangements.
Accumulating evidence suggests that in the case of RVD, which means that the cell decreases its volume from a swollen state after exposure to hypotonic stress, the immediate exit of inorganic ions, such as potassium and chloride, from the cell cytoplasm is triggered. Potassium and chloride ions are extruded through the cell membrane either by the activation of separate potassium and chloride channels or by the activation of potassium/chloride cotransporters1,2 (Figure 1). It has been shown experimentally that, in the case of cell swelling, the permeability of the cell membrane to chloride increases by sixtyfold, whereas the permeability to potassium increases only twofold. This means that chloride transport is the principal factor affecting the RVD adjustment.10
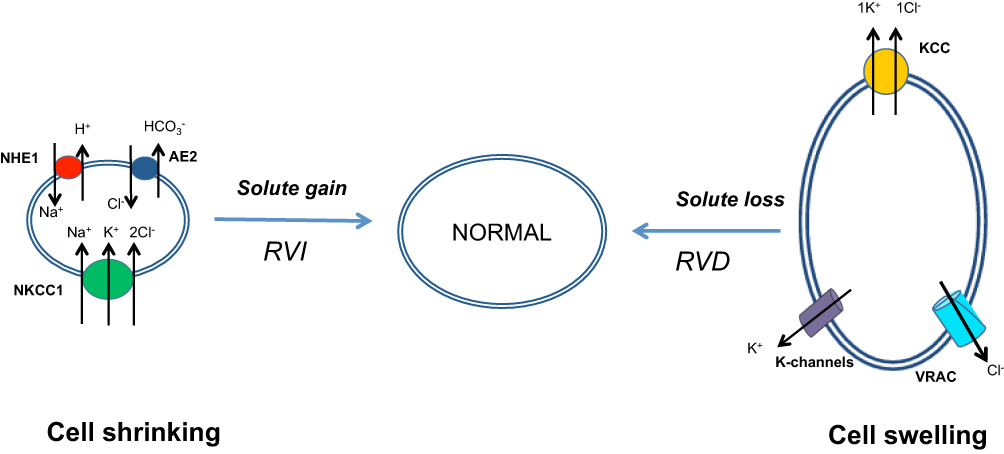 |
Figure 1 Cell volume regulation mechanisms. The regulatory volume increase is characterized by intracellular accumulation of osmotically active ions, mainly sodium, potassium, and chloride. The regulatory volume decrease is characterized by the extrusion of osmotically active ions, mainly potassium and chloride, outside the cell. Chloride anions play a crucial role in mediating the RVI and RVD. Abbreviations: RVI, Regulatory Volume Increase; RVD, Regulatory Volume Decrease; NHE1, Sodium Hydrogen Exchanger-1; AE2, Anion Exchanger-2; NKCC1, Sodium-Potassium-2 Chloride Cotransporter 1; KCC, Potassium-Chloride Cotransporter; VRAC, Volume Regulated Anion Channel; K-channels, Kv; Kca; Kirs; K2ps –channels. |
As early as 1978 and later in 1982, experimental evidence showed that RVD activates an outward rectifying chloride current (ICl,swell) in a time period of 4–6 min and is present in all vertebrate cell types examined.11 The underlying chloride channel was named volume-regulated anion channel (VRAC). It was recognized that, apart from chloride anions, VRAC carries outside the cell small organic osmolytes, mainly taurine, and constitutes one of the most effective RVD adaptive mechanisms.
Molecular identification of this channel was elusive until 2014, when two separate laboratories, Qiu Z et al12 and Voss FK et al13 identified and cloned an essential protein of this channel, SWELL1, which is a member of the five transmembrane proteins of the leucine-rich repeat (LRR)-containing domain and is the product of the LRRC8A gene. The LRRC8 family consists of five members, A to E. Further investigation showed that VRAC channels are formed as a result of heteromerization of the SWELL1 protein with at least one of the other four LRRC8 protein members (LRRC8B-E) of the family and form functioning hexamers. Certain participation of the other members ensure specific properties of the channel, such as volume sensing, specification in ion or organic osmolyte transport, and set point of activation.14
In the case of RVI adaptation, the cell volume increases after transient initial shrinking as a consequence of exposure to hypertonic stress. First, most cells attempt to manipulate the hypertonic osmotic gradient by losing intracellular water, and achieve osmotic equilibrium by shrinking. Under certain circumstances, they retain the capacity to increase their volume. For example, when placed in an isotonic environment, they eventually exhibit RVI to restore their prior volume.6
In general, cells manipulate RVI in two phases: the short-term RVI and long term. In the first case, cells accumulate inorganic ions, and if hypertonicity persists, some cells accumulate organic osmolytes, whereas others remain shrunk.6 As early as 1971, Kregenow15 showed that duck erythrocytes were placed in a hypertonic medium and regained their normal volume by intracellular movement of potassium and chloride ions after initial shrinking.
We now know that RVI triggers the intracellular accumulation of potassium chloride and sodium chloride. The movement of these ions is accomplished via activation of sodium/hydrogen exchanger 1 (NHE1), chloride/bicarbonate exchanger (Cl−/HCO3−) (AE2), and sodium-potassium-2 chloride cotransporter 1 (NKCC1).1,6,16 As depicted in Figure 1, cell shrinking activates NHE1, which senses intracellular osmolality or volume decrease via an elusive mechanism. This results in the increased intracellular movement of sodium ions (Na+) in exchange for hydrogen ions (H+). The intracellular pH increases as a consequence of a decrease in intracellular H+ concentration. Intracellular alkalinization causes an influx of carbon dioxide (CO2) and produces carbonic acid, which in turn increases the intracellular concentration of bicarbonate anions (HCO3−), resulting in an increase in Cl−/HCO3− exchanger activity.16 The coupled action of these two antiporters produces a net flux of Na+ and Cl− ions inside the cell. This increases intracellular osmolality and results in increased movement of water inside the cell. This type of restoration is especially encountered in erythrocyte volume regulation of most species examined including humans and is known as “Hamburger phenomenon” or “chloride shift” after Hartog Jacob Hamburger described it in 1919.17 NKCC1 and NHE1 are the main mediators of RVI, with the exception of red blood cells from some species such as teleost fish and amphibians. In these species, NKCC1 is not expressed, and RVI is accomplished mainly via the coupled action of NHE1 and chloride/bicarbonate exchanger.18
Cation-chloride cotransporters (CCCs) have also been implicated in cell volume regulation; however, they are the main carriers of inorganic ions. These ions facilitate entry into cells during short-term RVI restoration. The solute carrier-12A (SLC-12A) family of chloride cotransporters encompasses two subfamilies of electroneutral chloride cotransporters with sodium and/or potassium. The first subfamily has three members: sodium-potassium-2 chloride cotransporters (NKCC1 and NKCC2) and a sodium-chloride cotransporter (NCC). The second subfamily contains four members of the potassium chloride cotransporter (KCC1-4).18 Two isoforms of the first subfamily, NKCC1 and NKCC2, have been isolated from the vertebrate epithelia. NKCC1 is ubiquitously present in almost all species and is preferentially located in the basolateral membrane of the secretory epithelia. NKCC2 is found only in the thick ascending limb of Henle’s loop (TAL) and is expressed on the luminal surface of the cell membrane. Sodium, potassium, and chloride were carried inside the cell at a stoichiometry of 1:1:2. The NCC is located in the luminal cell membrane of the distal convoluted tubule, and carries sodium and chloride inside the cell at a stoichiometry of 1:1.18 KCCs represent another subfamily of electroneutral potassium/chloride cotransporters. They carry potassium and chloride at a stoichiometry of 1:1 outside the cell and are located in the basolateral cell membrane.18
Cell shrinkage and low intracellular chloride concentrations are the main factors that regulate the activity of these two subfamilies of cotransporters in the opposite direction. NKCCs phosphorylation leads to their activation, whereas KCCs phosphorylation leads to their inactivation.18 After rigorous experimental investigation, it became evident that the activation of NKCC1 and NKCC2 is accomplished via the phosphorylation of three specific threonine residues (Thr199, Thr201 and Thr206) in the NH2-terminus of the cotransporter molecule. In contrast, phosphorylation of Thr991 and Thr1048 at the carboxyl-terminus of KCCs can induce deactivation of the cotransporter molecule. This opposing action favors the accumulation of intracellular sodium, potassium, and chloride, which results in increased intracellular osmolality and water influx, resulting in an increase in intracellular volume. Both families of cotransporters are regulated by with-no-lysine kinases (for a detailed description refer to Chapter 3).19,20
Chloride anions and potassium cations predominate among the inorganic ions recruited for the immediate repair of cell volume disturbances, either RVD or RVI, signifying the fundamental role of the intracellular chloride concentration as an important regulator of cell volume.
Intracellular Chloride Homeostasis
Chloride is the most abundant anion in vertebrates because its principal role is to counteract and neutralize the positive currents of all other inorganic and organic cations. In humans, chloride anions represent approximately 70% of all anions, and the body of an average adult contains approximately 115 gr of chloride.17
The extracellular chloride concentration is relatively constant and is maintained between 95 and 120 mM among various species as a function of the kidney and intestine, which regulate its absorption and secretion. In humans, extracellular chloride concentration is maintained between 97 and 107 mM.17,21 The intracellular chloride concentration is maintained via complex and sophisticated manipulations. Even in the absence of active transport by the cell membrane, the intracellular chloride concentration is lower than that in the extracellular space due to Gibbs-Donnan disequilibrium. According to Gibbs-Donnan theory, cells contain non-diffusible organic anions that are osmotically active. The presence of these anions produces an electric gradient that pushes small anions, such as chloride, out of the cell membrane while favoring the retention of small cations, such as sodium and potassium, inside the cell. According to this assumption, cells are prone to swelling and contain lower amounts of chloride anions than the extracellular space.6
Intracellular chloride concentration varies greatly among different cell types in the same organism. Its concentration is dependent on the resting membrane potential and pH. In epithelial cells, the chloride concentration is approximately 40 mM, 30 mM in skeletal muscle, and 5 mM21,22 in most mature neurons. For further insights into the interaction between chloride channels and neuron function, please refer to the dedicated section below. Intracellular chloride homeostasis is a dynamic process accomplished via the coupled action of a plethora of transporters and ion channels, most of which are located in the cell membrane and others in the cytoplasm, as well as in the membrane of intracellular organelles.23,24
Chloride movement inside the cell is achieved via cation cotransporters (CCCs), mainly sodium and/or potassium, such as Na+/K+/2Cl− cotransporters (NKCC1-2) and Na+/Cl− cotransporters (NCC). These cotransporters use the sodium concentration gradient between the intracellular and extracellular spaces as the driving force for chloride movement. NKCC1 is ubiquitously present in most cells and primarily located in the basolateral membrane. NKCC2 is predominantly present on the apical (lumen) surface of epithelial cells in the thick ascending limb of the Henle’s loop (TAL). NCC is predominantly present on the lumen surface of epithelial cells in the distal convoluted tubule (DCT1) and is the main sodium transporter in the early segment of the distal nephron.23,25
Chloride movement outside the cell is mainly achieved via potassium/chloride co-transporters (KCCs), which are located at the basolateral membrane and drive potassium outside the cell along with chloride in a stoichiometry of 1:1. The driving force for these co-transporters is the concentration gradient of potassium between intracellular and extracellular spaces. Chloride concentration is greater in the extracellular space than in the intracellular space, and the opposite is true for potassium. Nevertheless, the potassium concentration gradient was greater than that of chloride. This predominance of the potassium gradient thermodynamically favors the movement of potassium and chloride via KCCs outside the cell. Four isoforms of these cotransporters (KCC1-4) which are distributed in many cell types and tissues, including red blood cells, vascular smooth muscle, epithelial cells, myocardium, skeletal muscle, auditory cells, and neurons. In kidney epithelial cells, KCC1, 3, and 4 are the most abundant, whereas KCC2 is predominant in CNS postsynaptic dendrites, co-localized with GABAA and glycine receptors.26,27
Another family of chloride channels that plays an important role in cell function and certain diseases are voltage-gated electrogenic CLC channels. These channels are widely distributed among all phyla and consist of nine members (CLC1-7) plus two kidney-specific members (Ks), CLC-Ka and CLC-Kb, which correspond to CLC-K1 and CLC-K2 encountered in rodents.23,25 The first member of the chloride channel family was purified and characterized in Torpedo Californica electroplax in 1990 by Jentsch et al. One year later, the same team purified and characterized another member of the family in the frog skeletal muscle and named it CLC1. Later, they decided to give the name CLC0 in the Torpedo electroplax channel so that it stands alone among this family members.23
The first four members of the chloride channel family (CLC1-2, CLC-Ka, and CLC-Kb) are primarily located in the cell membrane, whereas the other five members (CLC3-7) are located in the membranes of intracellular organelles (endoplasmic reticulum and lysosomal membranes). They are chloride-conducting channels, except CLC3-7, which act as chloride/hydrogen exchangers (2Cl− /1H+) with a stoichiometry of 2:1. The two kidney-specific members (CLC-Ka and CLC-Kb) require the presence of a beta subunit chaperone, barttin, to express their maximal activity and membrane localization.23,25
CLC1 is highly expressed in skeletal muscles and provides inward movement of chloride, which contributes to cell membrane repolarization immediately after depolarization and stabilizes the voltage of the cell membrane. CLC2 is widely expressed in epithelial and non-epithelial cells, such as in the brain, heart, and gastrointestinal tract. CLC-Ka and CLC-Kb are mainly expressed in the basolateral membrane of the renal epithelial cells, inner ear, and salivary glands.23
The activity of these channels is dependent on the cell membrane voltage and intracellular pH. Hyperpolarization of the cell membrane and intracellular alkalinization probabilistically increase the opening of the channels, favoring chloride exit outside the cell. The extracellular chloride concentration affects the activity of CLC2, as it has been proven experimentally that lowering the external chloride concentration activates chloride currents via CLC2.28 The direction of chloride transport is dependent on the electrochemical gradient of the chloride anions across the cell membrane. As a result, the opening of CLCs produces either an inward or outward chloride current, resulting in either hyperpolarization or depolarization of the cell membrane.23 These data suggest that intracellular chloride concentration plays a crucial role in determining intracellular osmolality, intracellular volume, and cell membrane potential, as well as the pH of the intracellular organelles.
Sodium Chloride Reabsorption in Distal Nephron: Role of WNKs and Extracellular Potassium
Sodium chloride reabsorption in the distal nephron is an important process for two reasons. First, this segment of the renal tubule is located beyond the macula densa; hence, it is outside the regulatory mechanism of the tubuloglomerular feedback. This means that the amount of sodium that escapes reabsorption in this segment of the nephron is lost in the urine and therefore poses a threat of extracellular volume depletion. Second, the late segments of the distal convoluted tubule (DCT), namely the DCT2, the connecting tubule (CNT), and the cortical collecting duct (CCD), represent the aldosterone-sensitive segment of the nephron, which achieves potassium homeostasis by reabsorption of sodium via the epithelial sodium channel (ENaC) and excretion of potassium via the renal outer medullary potassium channel (ROMK).29
The amount of sodium reabsorbed in this segment of the nephron is approximately 7% of the total sodium filtered in Bowman’s space; however, it is sufficient to produce considerable disturbances in the extracellular volume and blood pressure regulation if it is not properly conserved. Given that the daily amount of sodium filtered into the glomerulus is approximately 25,000 mmol the amount absorbed by DCT1 is 1750 mmol, which equals 40.25 gr of sodium. This is a considerable amount of sodium and is very difficult to replace by food if it were to be lost in the urine.5
The distal convoluted tubule (DCT) is characterized by the expression of the sodium chloride cotransporter (NCC) on its luminal membrane, exclusively in DCT1. In the early segment of DCT2, there is co-expression of NCC and the epithelial sodium channel (ENaC), while ENaC becomes the sole sodium transporter on the luminal surface of DCT2. Sodium transport within the cell is electrogenic, and the activity of ENaC is counteracted by the concomitant activation of the renal outer medullary potassium channel (ROMK). ROMK, a potassium channel, drives potassium ions to the tubular lumen, establishing luminal negativity that promotes potassium excretion and facilitates chloride reabsorption. Both ENaC and ROMK are regulated by aldosterone. Thus, the tubular excretion of potassium via ROMK, along with the voltage-sensitive calcium-activated big potassium channel (BK-channel), represent the primary mechanisms of potassium excretion in the DCT.29
It is obvious that the DCT represents the segment of the nephron that ensures the fine-tuning of sodium balance and potassium excretion. Accumulated evidence from the study of inherited disorders in humans affecting this segment of the nephron, such as Gitelman syndrome and Familial Hyperkalemic Hypertension (FHHt), suggests that cases of ineffective function of this segment present a phenotype of low blood pressure accompanied by hypokalemia, whereas cases of a gain-of-function phenotype present with hypertension and hyperkalemia.29
It seems likely that the two segments of the DCT (DCT1 and CNT) communicate with each other and regulate their activities according to the demands of sodium reabsorption and potassium excretion. In cases of excessive extracellular potassium, NCC slows down its activity and increases the delivery of solutes to the CNT, where ENaC reabsorbs sodium with concomitant excretion of potassium via ROMK. An inverse sequence of events occurs under opposing conditions. It is well known that an increase in extracellular potassium concentration decreases the NCC activity and increases aldosterone production.28
Population-based epidemiological and experimental studies in animals have shown an inverse relationship between potassium intake and blood pressure. An increase in potassium intake results in a reduction in blood pressure, even among individuals with a high salt intake. A recent meta-analysis of 32 RCTs showed a U-shaped relationship between potassium intake and blood pressure measurement, indicating that excessive potassium intake (>100 mEq/day) may increase blood pressure.28,30
Until the beginning of the new millennium, our knowledge of the complex mechanisms that regulate sodium chloride and potassium homeostasis in the distal nephron was elusive. In 2000 Bing-e Xu et al31 discovered a new protein kinase, named with-no-lysine Kinase 1 (WNK1). The name was derived from the fact that the new kinase lacks the characteristic catalytic lysine of all other known protein kinases in position 72 (Lys72) in β-strand 3 and instead possesses its catalytic lysine at position 233 (Lys233) in β-strand 2. It became evident from the beginning of its discovery that WNK1 is capable of auto-phosphorylation under hypertonic conditions produced by increased concentrations of NaCl or mannitol.31
Subsequent investigations have shown that the family encompasses four members, WNK1 to WNK4, which act as serine/threonine kinases. They are conserved in many species and widely distributed in many cell types. In humans, WNK2 is distributed almost exclusively in the central nervous system. WNK3 is distributed in many cell types and is expressed throughout the renal epithelia; however, its expression is scant in DCT. WNK4 is expressed in many cell types and in renal epithelial cells, such as the thick ascending limb of Henle’s loop (TAL), DCT, and CCD. WNK1 is ubiquitously expressed in almost all cell types. The kidneys express at least two isoforms of the kinase: the long isoform, which represents the long molecule of the kinase (L-WNK1), and a specific short isoform known as KS-WNK1, which is expressed predominantly in the DCT. This isoform is the product of an alternative promoter located in intron 4a, and starts transcription from the late part of intron 4, continuing to the rest of the molecule from intron 5. As a result, the KS-WNK1 molecule contains an N-terminal consisting of only 30 residues and lacks the characteristic catalytic domain; thus, it is inactive as a kinase.3,29
WNK1 is the most extensively studied molecule in this family; however, all its members share common structural motifs with characteristic functions. For example, the kinase domain is located at the N-terminus (residues 218–483) and two coiled-coil domains at the C-terminus (residues 563–597 and 1814–1841). They also possess an auto-inhibitory domain that suppresses kinase activity (residues 490–550); which lies in close proximity to the kinase domain, and contains at least two phenylalanine residues. WNKs exhibit autophosphorylation capacity at serine 382 (Ser382), with resultant activation. Another autophosphorylation site is detected in serine 378 (Ser378). Phosphorylation of Ser382 activated kinase molecules by 100%, whereas phosphorylation of Ser378 activated kinase molecules by only 50%. Consequently, phosphorylation of S382 represents the active form of WNK kinases.3,32
Downstream substrates of WNKs are two other serine/threonine kinases, namely Ste-20 proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase-1 (OSR1). A detailed investigation of this process by Vitari et al33 showed that activated WNKs activate SPAK by phosphorylation of Thr233 and OSR1 by phosphorylation of Thr185 in the T-loop domain, as well as a specific serine residue in the C-terminal domain, Ser373 in SPAK, and Ser325 in OSR1.33,34 Activated SPAK and OSR1 kinases phosphorylate and activate NCC. In contrast, they phosphorylate and deactivate the KCCs. The opposite action of phosphorylation aims to increase the intracellular chloride concentration. Phosphorylation of three amino acid residues in the N-terminal domain of the NCC molecule activates the cotransporter. The phosphorylation of threonine 53 (T53), threonine 58 (T58), and serine 71 (S71) is essential for transporter activation. In contrast, phosphorylation of KCC at threonine 991 (T991) and threonine 1048 (T1048) deactivates the cotransporter, whereas dephosphorylation activates the molecule.19,34
In 2014, Piala et al35 showed experimentally that WNK1 autophosphorylation and activation are dependent on intracellular chloride concentration. The WNK1 molecule contains a specialized structure known as the DLG motif, which is composed of certain amino acids such as Phe283, Leu299, Leu369 and Leu371. This motif can form hydrophobic chloride-hydrogen bonds with the chloride anions. The trapping of chloride anions in the DLG motif produces conformational changes in the WNK1 molecule, which inhibit Ser382 phosphorylation and deactivate the kinase. In cases of increased intracellular chloride concentration, autophosphorylation and activation of the WNK1 molecule are prevented, whereas in cases of decreased intracellular chloride concentration, chloride anions are released from the DLG motif, and autophosphorylation and activation of WNK1 is feasible. According to these findings, WNK1 acts as an intracellular chloride sensor that modulates its activity according to intracellular chloride concentration (Figure 2). These data clearly indicate that NCC activity is mainly regulated by the WNK/SPAK/OSR1 kinase cascade activity, which in turn is regulated by intracellular chloride concentration. The question is about the identity of the molecule that determines the activity of this cascade, and activates or deactivates this biological system on demand. Given the fundamental role of the kidney in extracellular volume conservation by regulating sodium balance, this role could be attributed to sodium; however, the aldosterone paradox indicates that potassium homeostasis overrides volume disturbances, and therefore a potential candidate for this role could be potassium.
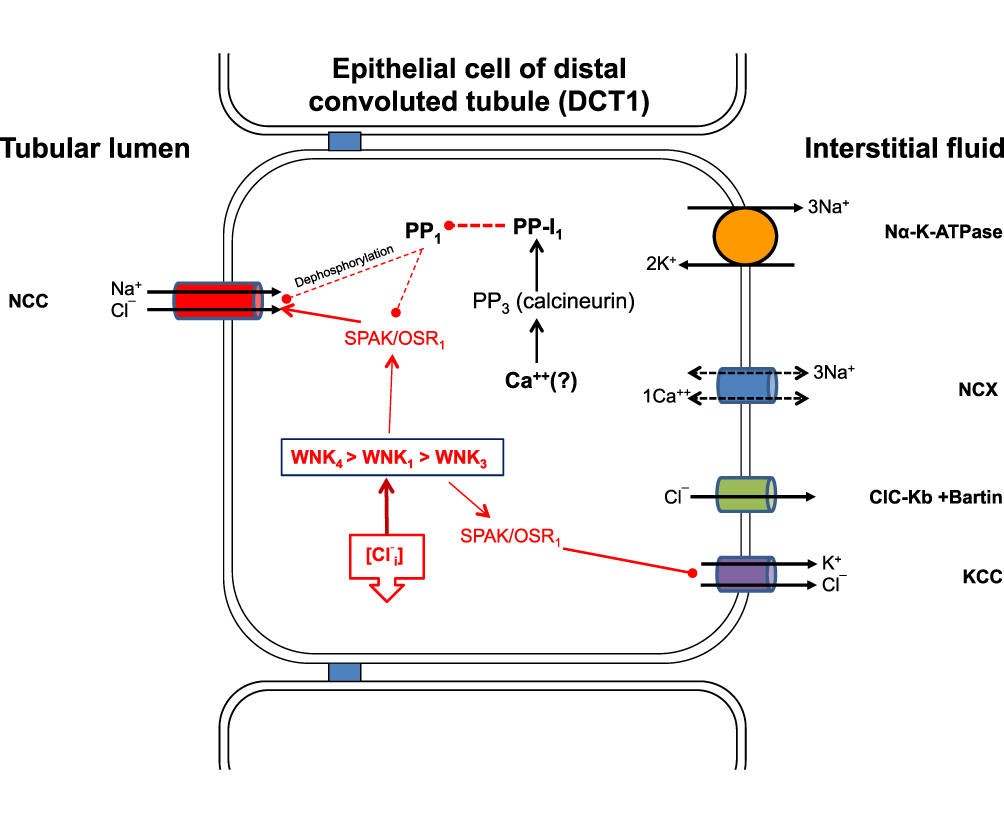 |
Figure 2 Solute transport in the DCT1. The sodium/chloride cotransporter is the only sodium chloride cotransporter found in the luminal membranes of epithelial cells. Phosphorylation and subsequent activation of the transporter are achieved via the WNKs/SPAK/OSR1 cascade, which in turn is activated by the reduction of intracellular chloride concentration. The inactivation of NCC by dephosphorylation is partially dependent on intracellular chloride concentrations. Sufficient evidence suggests that increased levels of extracellular potassium lead to rapid dephosphorylation of NCC via an alternative metabolic pathway that encompasses the protein phosphatase family PP1-PP4 and protein phosphatase inhibitor-1. This pathway is not well understood, but intracellular calcium and calcineurin appear to play crucial roles. Abbreviations: NCC, Sodium/chloride cotransporter; WNK, With-no-lysine kinase; SPAK, Ste-20 Proline-alanine rich kinase; OSR1, Oxidative stress responsive element-1; [Cl−i], Intracellular chloride; KCC, Potassium/chloride cotransporter; CLC-Kb+Barttin, Chloride channel Kb+Barttin; NCX, Sodium/Calcium exchanger; PP1, Protein phosphatase 1; PP-I1, Protein phosphatase Inhibitor 1. |
In a detailed experimental study, Terker et al36 showed that a low-potassium diet activates NCC and, under conditions of a high-salt diet, increases blood pressure. Conversely, a normal potassium diet deactivates NCC and, even under high-salt diet conditions, reduces blood pressure. They showed that potassium diet affects the extracellular potassium concentration, which in turn affects the potential of the basolateral membrane of DCT cells. Specifically, a low-potassium diet reduces extracellular potassium levels, resulting in the efflux of potassium from the intracellular to the extracellular space. This movement of potassium causes hyperpolarization of the cell membrane and decreases the intracellular chloride concentration, resulting in the activation of the WNK/SPAK/OSR1 cascade and activation of NCC.
The main potassium channels in the basolateral membrane of DCT cells are two members of the inwardly rectifying potassium channels (Kirs), namely Kir4.1 and Kir5.1). Kir4.1, which is the product of the KCNJ10 gene, and Kir5.1, which is the product of the KCNJ16 gene. They were expressed in abundance in the TAL, DCT, CNT, and CCD. These two channels are capable of forming a functional heterotetramer with a 40 pS K+ conductance channel that drives potassium outside the cell.37,38 The first member of this family was characterized as “anomalous” rectifying potassium channel because it facilitates the inward movement of potassium against its concentration gradient. The direction of potassium movement depends on the membrane localization of the channel (apical or basolateral), extracellular potassium concentration, and cell membrane potential.39
Experimental studies in mice showed that a low-potassium diet activates the Kir4.1 channel and increases potassium exit outside the DCT basolateral cell membrane, resulting in membrane hyperpolarization, whereas a high-potassium diet has the opposite effect. Membrane hyperpolarization results in an increased chloride current outside the cell because of activation of the chloride channel CLC-Kb. This is the main chloride channel in the basolateral membrane of the DCT, and its activation decreases intracellular chloride concentration. This results in the activation of the WNK/SPAK/OSR1/NCC axis and increases NCC phosphorylation (pNCC). Deletion of Kir4.1 in kidney-specific-Kir4.1 knock out mice (KS-Kir4.1 KO) abolished potassium and chloride currents, as well as hyperpolarization of the cell membrane during a low-potassium diet29,36–38 (Figure 3). Under a high-potassium diet, extracellular potassium concentration increases, leading to NCC dephosphorylation. Experimental studies in animals have shown that potassium loading produces acute natriuresis in 30–60 minutes and kaliuresis for different durations. Kaliuresis persisted for approximately 6 h, whereas natriuresis decreased after approximately 3 h. The same effect of potassium loading has been confirmed in humans. Experimental studies in mice under low, normal, and high-sodium diets, when challenged with an oral potassium load (as KCl), showed NCC dephosphorylation even in a high-salt diet, pointing to the possibility that potassium homeostasis overrides volume homeostasis.40,41
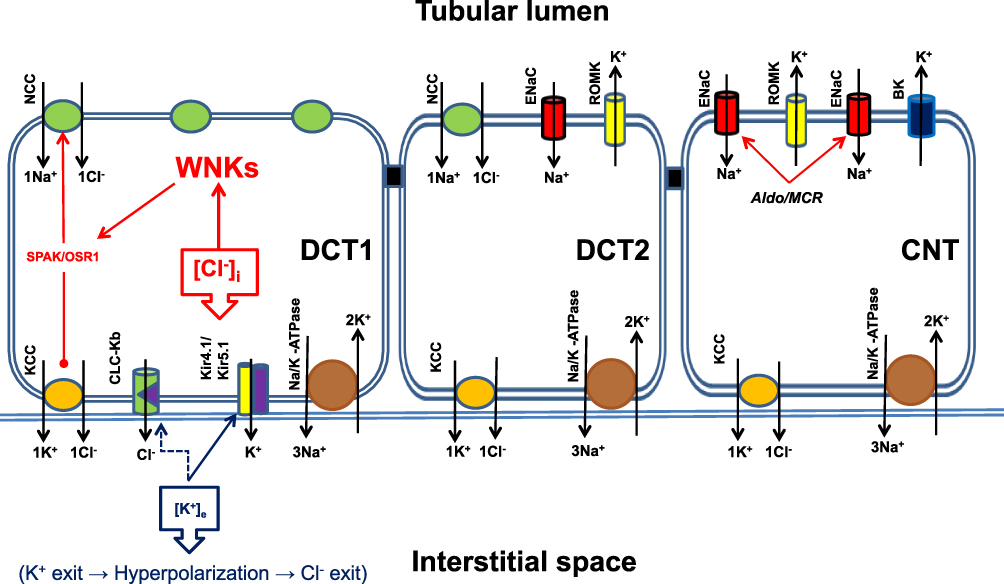 |
Figure 3 Solute Transport in the distal nephron. The main sodium carrier in the early segment of the distal nephron (DCT1) is the sodium/chloride cotransporter NCC, which is almost exclusively expressed in this segment of the nephron. Next, NCC was expressed in the first portion of DCT2 and gradually replaced by ENaC. In the connecting tubule (CNT), only ENaC is expressed, accompanied by the potassium-excreting channels ROMC and BK-channels. The potassium excretion capacity of this nephron segment depends on the amount of solute and sodium delivered by DCT1 and the presence of aldosterone. The activity of NCC is regulated by WNK kinases, which are regulated by the intracellular chloride concentration. Low intracellular chloride concentrations increase WNKs activity, and hence NCC activity. Extracellular potassium concentration is sensed by DCT1 via the inwardly rectifying potassium channel Kir4.1 located at the basolateral membrane of DCT1. A low extracellular potassium concentration activates heterodimers of Kir4.1/Kir5.1 potassium channels and increases the efflux of intracellular potassium to the extracellular space, which produces membrane hyperpolarization and activation of the chloride channel CLC-Kb. Chloride efflux reduces intracellular chloride concentration and activates WNKs, which in turn activates NCC and increases NaCl reabsorption by this segment of the nephron, consequently reducing sodium delivery to DCT1 and CNT. This sequence of events favors the reduction of potassium excretion by the distal nephron to conserve the extracellular potassium balance. Opposite events occurred in patients with increased extracellular potassium concentrations. Abbreviations: NCC, Sodium/chloride cotransporter; KCC, Potassium/chloride cotransporter; CLC-Kb, Chloride channel Kb + Barttin; Kir4.1, Inwardly rectifying potassium channel 4.1; Kir5.1, Inwardly rectifying potassium channel 5.1; ENaC, Epithelial sodium channel; ROMK, Renal outer medullary potassium channel; BK, Big potassium channel; Aldo/MCR, Aldosterone + Mineral corticoid receptor; [Cl−]I, Intracellular chloride concentration; [K+]e, Extracellular potassium concentration. |
Accumulated evidence from experimental studies suggests that NCC dephosphorylation by plasma potassium is feasible in the range of normal plasma potassium concentrations (3,0–5,0 mM), and an increase of 0.1 mM of extracellular potassium concentration produces a decrease in pNCC by approximately 15%.41 The mechanism by which extracellular potassium decreases NCC phosphorylation appears independent of intracellular chloride changes. Penton et al40 showed experimentally that extracellular potassium concentration increments, within the normal variations of plasma potassium, are capable of rapid NCC dephosphorylation, within 5–15 minutes, which is partially independent of the Cl/WNKs/SPAK/OSR1 pathway and may encompass other mediators, such as the activation of intracellular phosphatases, which leads to rapid NCC dephosphorylation.
Based on recent experimental data, it seems likely that an increased extracellular potassium concentration affects NCC activity via different metabolic pathways. The first pathway was the Cl/WNK/SPAK-OSR1 pathway. The second pathway involves the activation of intracellular kinases that activate intracellular protein phosphatases (PP), which in turn dephosphorylate and deactivate NCC.29,41 It seems likely that intracellular PPs participating in NCC dephosphorylation are PP1, PP2, PP3 (calcineurin), and PP4. Evidence from experimental studies suggests a role for these phosphatases. On the other hand, it is well known that immunosuppressive drugs such as tacrolimus and cyclosporine, known inhibitors of calcineurin, are used in transplantation and autoimmune disease therapy and are responsible for hypertension and hyperkalemia among these patients.29,41 How the increased extracellular potassium concentration decreases pNCC via this pathway is still not well understood. Experimental findings have shown that calmodulin and calcineurin can dephosphorylate NCC. These proteins are sensitive to intracellular calcium concentrations. Therefore, a possible explanation is that an increased extracellular potassium concentration leads to membrane depolarization, which results in the opening of voltage-sensitive Ca++ channels and increased intracellular Ca++ concentration, with subsequent activation of calmodulin/calcineurin and PPs. However, this assumption was not experimentally confirmed. More recent data suggest that WNKs are capable of sensing intracellular potassium concentrations, and cases of increased intracellular potassium lead to inhibition of kinase activity, indicating that intracellular chloride and potassium levels exert an additive effect on WNKs deactivation.41,42
Another issue that is not yet well defined is the capacity of WNKs to autophosphorylate and activate under hyperosmotic stress conditions. Recent experimental data suggest that, under normal circumstances, WNK1 and WNK3 exist in a dimerized form, which is capable of attracting water molecules and representing a more hydrated conformational state. This hydrated form is characterized by the exposure of the inhibitory site, which is occupied by chloride, and the molecule is unphosphorylated and inactivated. Under hyperosmolar stress, water molecules are separated from this hydrated state and WNKs dissociate into monomers. Subsequently, the chloride anion was removed from the inhibitory site and the monomers were activated via autophosphorylation.43
Chloride Channels and Neuron Function
Intracellular chloride concentration plays a crucial role in normal neuronal development and function. Mature neurons typically maintain a lower intracellular chloride level, approximately 5 mM. The KCC2 transporter primarily drives chloride out of neuronal cells, while the NKCC1 transporter facilitates the movement of chloride into the cells. The balance between these transporters determines the intracellular chloride concentration in neurons.
During neuronal excitation, signals are rapidly propagated to postsynaptic dendrites, leading to the excitation of neighboring neurons. If this excitation is not confined to a restricted area, it can spread extensively in the brain and result in epileptic seizures. Following neuronal excitation, GABAA and glycine receptors on the postsynaptic dendrites are activated. This activation allows for rapid intracellular chloride influx through receptor-activated chloride channels, leading to a significant increase in intracellular chloride levels, reaching up to 20 mM within a few hundred milliseconds.
The rapid rise in intracellular chloride induces hyperpolarization and stabilizes postsynaptic neurons, preventing the spread of excitation. This influx of extracellular chloride into the postsynaptic dendrites is facilitated by the low levels of intracellular chloride. Dysfunctions in the KCC2 transporter or an imbalance in the function between NKCC1 and KCC2 have been associated with childhood epilepsy, certain autism spectrum disorders, and chronic neuropathic pain.22
An Integrative Approach
Kidneys play a crucial role in extracellular volume regulation and composition of the internal milieu. This process is accomplished by a delicate and rigorous function of the nephron, which is capable of filtration, reabsorption, and excretion of inorganic ions and organic molecules in such a manner that maintains fine regulation of body composition, which sustains survival in terrestrial life.44
The above process is accomplished by the daily production of approximately 180 L of plasma ultrafiltrate in Bowman’s space, which has an identical composition to plasma, except for proteins with molecular masses greater than 30.000 Daltons. The final amount of urine excreted by the kidneys approaches 1% of the initial ultrafiltrate, approximately 1.5 L per day. The amount of daily sodium, potassium, and chloride filtered through the glomerulus was approximately 24,000 mmol for sodium, 600 mmol for potassium, and 16,000 mmol for chloride. Although most of these ions are reabsorbed by the paracellular pathway, a considerable amount of solutes are reabsorbed by the transcellular pathway, especially in TAL, DCT, and CCD.5,45
Nephron epithelial cells must reabsorb and excrete large amounts of solutes on a diurnal basis. Most solutes are transferred through the cell body and produce dynamic changes in intracellular osmolality, which must be immediately restored to ensure cell survival and proper function. In fact changes in osmolality, though not as strong initially, are evident even from the proximal convoluted tubule (PCT) where micropuncture studies have shown a difference of approximately 7 mOsm/kg H2O between the tubular lumen and interstitial space. While it is true that the majority of solute transport in the proximal tubule occurs via the drag phenomenon or passive diffusion through the cell body, the downstream nephron segments exhibit dynamic changes in intracellular osmolyte concentration. These changes in osmolality are particularly significant in the distal convoluted tubule (DCT) and connecting tubule (CNT). Furthermore, the countercurrent multiplication system of the kidney establishes an osmolality difference, known as the “single effect” of urine concentration, between the thin descending and thin ascending limbs of the loop of Henle.46 This osmolality difference, estimated to be around 20 mOsm/kg H2O, contributes to the subsequent concentration of urine in the distal parts of the nephron. These findings highlight the dynamic nature of intracellular osmolality regulation and indicate that there are indeed changes in osmolality between the tubular fluid and interstitial space in the DCT and CNT.
Reclamation of the filtered load of water and solutes in Bowman’s space is achieved in varying amounts across the nephrons. The proximal tubule reabsorbs approximately 60% of the filtered water and sodium, another 25% is reabsorbed in the TAL, approximately 10–15% in the distal nephron, and the remainder 1–2% is reabsorbed in the collecting duct. Potassium reabsorption across nephrons is more complex than sodium reabsorption. Approximately 60% of filtered potassium is reabsorbed in the proximal tubule, another 25% is reabsorbed in the TAL, and the remainder is excreted in the distal nephron, mainly coupled with sodium reabsorption.5,45
According to this scheme, the proximal nephron is dedicated to conserving the vast majority of filtered sodium and water, the TAL is used to reabsorb the amount of sodium needed to conserve the hypertonicity of the renal medulla, and the distal nephron is used to fine-tune sodium reabsorption and excrete excess potassium.
Human dietary habits have changed dramatically over the past 300 generations. Paleolithic humans consumed a diet rich in potassium and poor in sodium. According to data collected from studies exploring the suggested composition of the Paleolithic diet, humans consumed 20–40 mM of sodium per day and potassium ranged between 400 and 500 mM per day (ratio of K+ /Na+ = 10 to 12.5). Conversely, modern diets in industrialized communities with processed foods consist of high sodium and low potassium levels. Contemporary diets contain approximately 100–400 mM sodium per day and 30–70 mM potassium (K+ /Na+ = 0.3 to 0.17).47,48 The evolutionary adaptation of our genome over the past to 6–7 million years has provided our ancestors with the capacity to fully accommodate their dietary habits. Contemporary dietary habits have gradually changed since the evolution of agriculture approximately 10 thousand years ago. This period is very short on an evolutionary scale to produce any significant genome adaptations capable of accommodating contemporary dietary habits. According to this assumption, the kidneys of modern humans are genetically engineered to efficiently accommodate Paleolithic diets and, as a consequence, are capable of performing rigorous reabsorption of sodium and excrete a huge amount of potassium.49,50
In the modern era, the ratio of ingested K+ /Na+ has diminished dramatically, and as our kidneys remain adapted to Paleolithic diets, they continue to reabsorb sodium, but they also have to conserve potassium. Potassium excretion in the distal nephron is coupled with sodium reabsorption and in cases of low potassium ingestion, as is the rationale for contemporary diets; nephrons have to diminish the amount of solute and sodium delivered from the DCT1 to CN tubule and CCD to suspend potassium excretion in this segment of the nephron. This goal is achieved by increasing the activity of NCC, which reabsorbs NaCl, resulting in an increase in extracellular volume and blood pressure. This rigorous effort of nephrons to regulate extracellular potassium concentration seems likely to represent the underlying mechanism of sodium sensitivity, leading to sodium-sensitive hypertension.29,51
In contrast, when dietary potassium ingestion increases, the amount of potassium absorbed by the gastrointestinal tract has to be manipulated immediately by the kidney because the total amount of potassium distributed in the extracellular space is approximately 65 mM. This means that even contemporary diets, with an average potassium ingestion of about 30–70 mM per day, are capable of doubling the amount of extracellular potassium, which may result in dangerous and/or fatal hyperkalemia. Under these conditions, the organism immediately distributes potassium to the intracellular space; however, the final regulation of ingested potassium is achieved via excretion by the distal nephron. This vital goal is achieved with increased production of aldosterone and decreased activity of the NCC, which results in increased delivery of sodium chloride to the CNT and CCD when aldosterone excretes potassium and reabsorbs sodium. However, the amount of sodium that escapes reabsorption via ENaC in this segment of the nephron is high enough to produce natriuresis. It seems likely that this sequence of events sufficiently explains the hypotensive action of K-rich diets, even in the setting of increased sodium consumption and the aldosterone paradox observed in hyperkalemia.29,45,51
Conclusion
Chloride anion is one of the most important elements in the human body and its role is not only to neutralize the extracellular and intracellular cations. It actually contributes significantly to the osmolality of extracellular and intracellular compartments, thereby participating in the regulation of volume homeostasis. The integral role of chloride anion in cell volume regulation and hypertension is a complex and multifaceted phenomenon.
In the context of cell volume regulation, chloride anion serves as a crucial rescue osmole, particularly in regulatory volume decrease (RVD). It functions as the main osmole extruded outside the cell via volume-regulated anion channels (VRAC), rapidly reducing intracellular osmolality and restoring cell volume to normal.
Importantly, intracellular chloride plays a fascinating role in the regulation of With-No-lysine kinases (WNKs) through its capacity to regulate WNKs’ autophosphorylation and activation. The WNKs/SPAK metabolic pathway, regulated by intracellular chloride, further modulates the function of sodium-potassium-chloride cotransporters such as NKCC2, NCC, and KCC. This latter intricate interplay contributes to the regulation of salt reabsorption in the distal nephron, influencing renal medulla hypertonicity and sodium balance in the aldosterone-sensitive distal nephron. The regulation of NCC activity by intracellular chloride is crucial for potassium excretion in the aldosterone-sensitive distal nephron, controlling the solute and sodium delivery downstream to the nephron segments where aldosterone exerts its action. Extracellular potassium also participates in the modulation of NCC activity through chloride-dependent and chloride-independent pathways.
The coordinated action of intracellular chloride and extracellular potassium appears to underlie salt sensitivity in hypertension and the aldosterone paradox observed in hyperkalemia. Additionally, this may provide an explanation for the beneficial effects of high potassium-containing foods in ameliorating high blood pressure, even in individuals with high salt consumption.
In summary, chloride anion can be characterized as the “Queen of the game” in the intricate chessboard of renal physiology. Its involvement in cell volume regulation and its impact on various renal processes highlight its integral role in maintaining homeostasis.
Funding
The authors did not receive any funding, including financial support for the publication of this manuscript. This manuscript was created with authors’ personal funding.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28(1–4):155–159. doi:10.1152/advan.00034.2004
2. McManus ML, Churchwell KB, Strange K. Regulation of cell volume in health and disease. N Engl J Med. 1995;333(19):1260–1266. doi:10.1056/NEJM199511093331906
3. McCormick JA, Yang CL, Ellison DH. WNK kinases and renal sodium transport in health and disease: an integrated view. Hypertension. 2008;51(3):588–596. doi:10.1161/HYPERTENSIONAHA.107.103788
4. Orlov SN, Koltsova SV, Kapilevich LV, Gusakova SV, Dulin NO. NKCC1 and NKCC2: the pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015;2(2):186–196. doi:10.1016/j.gendis.2015.02.007
5. Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol. 2015;10(4):676–687. doi:10.2215/CJN.12391213
6. Delpire E, Gagnon KB. Water homeostasis and cell volume maintenance and regulation. Curr Top Membr. 2018;81:3–52.
7. De Smet P, Simaels J, Declercq PE, Van Driessche W. Regulatory volume decrease in cultured kidney cells (A6): role of amino acids. J Gen Physiol. 1995;106(3):525–542. doi:10.1085/jgp.106.3.525
8. Lopez-Cayuqueo KI, Planells-Cases R, Pietzke M, et al. Renal Deletion of LRRC8/VRAC channels induces proximal tubulopathy. J Am Soc Nephrol. 2022;33(8):1528–1545. doi:10.1681/ASN.2021111458
9. Baturina GS, Katkova LE, Schmitt CP, Solenov EI, Zarogiannis SG. Comparison of isotonic activation of cell volume regulation in rat peritoneal mesothelial cells and in kidney outer medullary collecting duct principal cells. Biomolecules. 2021;11(10):1452. doi:10.3390/biom11101452
10. Hoffmann EK, Simonsen LO. Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev. 1989;69(2):315–382. doi:10.1152/physrev.1989.69.2.315
11. Sardini A, Amey JS, Weylandt KH, Nobles M, Valverde MA, Higgins CF. Cell volume regulation and swelling-activated chloride channels. Biochim Biophys Acta. 2003;1618(2):153–162. doi:10.1016/j.bbamem.2003.10.008
12. Qiu Z, Dubin AE, Mathur J, et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 2014;157(2):447–458. doi:10.1016/j.cell.2014.03.024
13. Voss FK, Ullrich F, Munch J, et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 2014;344(6184):634–638. doi:10.1126/science.1252826
14. Konig B, Stauber T. Biophysics and structure-function relationships of LRRC8-formed volume-regulated anion channels. Biophys J. 2019;116(7):1185–1193. doi:10.1016/j.bpj.2019.02.014
15. Kregenow FM. The response of duck erythrocytes to hypertonic media. Further evidence for a volume-controlling mechanism. J Gen Physiol. 1971;58(4):396–412. doi:10.1085/jgp.58.4.396
16. Valles PG, Bocanegra V, Gil Lorenzo A, Costantino VV. Physiological functions and regulation of the Na+/H+ Exchanger [NHE1] in renal tubule epithelial cells. Kidney Blood Press Res. 2015;40(5):452–466. doi:10.1159/000368521
17. Astapenko D, Navratil P, Pouska J, Cerny V. Clinical physiology aspects of chloremia in fluid therapy: a systematic review. Perioper Med. 2020;9(1):40. doi:10.1186/s13741-020-00171-3
18. Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89(1):193–277. doi:10.1152/physrev.00037.2007
19. Rinehart J, Maksimova YD, Tanis JE, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138(3):525–536. doi:10.1016/j.cell.2009.05.031
20. Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK kinase signaling in ion homeostasis and human disease. Cell Metab. 2017;25(2):285–299. doi:10.1016/j.cmet.2017.01.007
21. Luscher BP, Vachel L, Ohana E, Muallem S. Cl(-) as a bona fide signaling ion. Am J Physiol Cell Physiol. 2020;318(1):C125–C136. doi:10.1152/ajpcell.00354.2019
22. Doyon N, Vinay L, Prescott SA, De Koninck Y. Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron. 2016;89(6):1157–1172. doi:10.1016/j.neuron.2016.02.030
23. Jentsch TJ, Pusch M. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev. 2018;98(3):1493–1590. doi:10.1152/physrev.00047.2017
24. Martinez AH, Mohiuddin SS. Biochemistry, Chloride Channels. In: StatPearls. Treasure Island (FL): StatPearls; 2022.
25. Teulon J, Planelles G, Sepulveda FV, Andrini O, Lourdel S, Paulais M. Renal chloride channels in relation to sodium chloride transport. Compr Physiol. 2018;9(1):301–342.
26. Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85(2):423–493. doi:10.1152/physrev.00011.2004
27. Garneau AP, Marcoux AA, Slimani S, et al. Physiological roles and molecular mechanisms of K(+) -Cl(-) cotransport in the mammalian kidney and cardiovascular system: where are we? J Physiol. 2019;597(6):1451–1465. doi:10.1113/JP276807
28. Pusch M, Jordt SE, Stein V, Jentsch TJ. Chloride dependence of hyperpolarization-activated chloride channel gates. J Physiol. 1999;515:341–353. doi:10.1111/j.1469-7793.1999.341ac.x
29. Castaneda-Bueno M, Ellison DH, Gamba G. Molecular mechanisms for the modulation of blood pressure and potassium homeostasis by the distal convoluted tubule. EMBO Mol Med. 2022;14(2):e14273. doi:10.15252/emmm.202114273
30. Filippini T, Naska A, Kasdagli MI, et al. Potassium intake and blood pressure: a dose-response meta-analysis of randomized controlled trials. J Am Heart Assoc. 2020;9(12):e015719. doi:10.1161/JAHA.119.015719
31. Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275(22):16795–16801. doi:10.1074/jbc.275.22.16795
32. Jonniya NA, Sk MF, Kar P. Investigating phosphorylation-induced conformational changes in WNK1 kinase by molecular dynamics simulations. ACS Omega. 2019;4(17):17404–17416. doi:10.1021/acsomega.9b02187
33. Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391(Pt 1):17–24. doi:10.1042/BJ20051180
34. Pacheco-Alvarez D, Cristobal PS, Meade P, et al. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281(39):28755–28763. doi:10.1074/jbc.M603773200
35. Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7(324):ra41. doi:10.1126/scisignal.2005050
36. Terker AS, Zhang C, McCormick JA, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21(1):39–50. doi:10.1016/j.cmet.2014.12.006
37. Cuevas CA, Su XT, Wang MX, et al. Potassium sensing by renal distal tubules requires kir4.1. J Am Soc Nephrol. 2017;28(6):1814–1825. doi:10.1681/ASN.2016090935
38. Wang MX, Cuevas CA, Su XT, et al. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018;93(4):893–902. doi:10.1016/j.kint.2017.10.023
39. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90(1):291–366. doi:10.1152/physrev.00021.2009
40. Penton D, Czogalla J, Wengi A, et al. Extracellular K(+) rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl(-) -dependent and independent mechanisms. J Physiol. 2016;594(21):6319–6331. doi:10.1113/JP272504
41. Hoorn EJ, Gritter M, Cuevas CA, Fenton RA. Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev. 2020;100(1):321–356. doi:10.1152/physrev.00044.2018
42. Pleinis JM, Norrell L, Akella R, et al. WNKs are potassium-sensitive kinases. Am J Physiol Cell Physiol. 2021;320(5):C703–C721. doi:10.1152/ajpcell.00456.2020
43. Akella R, Humphreys JM, Sekulski K, et al. Osmosensing by WNK Kinases. Mol Biol Cell. 2021;32(18):1614–1623. doi:10.1091/mbc.E20-01-0089
44. Hoenig MP, Zeidel ML. Homeostasis, the milieu interieur, and the wisdom of the nephron. Clin J Am Soc Nephrol. 2014;9(7):1272–1281. doi:10.2215/CJN.08860813
45. Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol. 2015;10(6):1050–1060. doi:10.2215/CJN.08580813
46. Sands JM, Layton HE. The physiology of urinary concentration: an update. Semin Nephrol. 2009;29(3):178–195. doi:10.1016/j.semnephrol.2009.03.008
47. Eaton SB, Eaton SB, Konner MJ. Paleolithic nutrition revisited: a twelve-year retrospective on its nature and implications. Eur J Clin Nutr. 1997;51(4):207–216. doi:10.1038/sj.ejcn.1600389
48. Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342(20):1493–1499. doi:10.1056/NEJM200005183422006
49. Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC
50. Roberts WC. Facts and ideas from anywhere. Proc Bayl Univ Med Cent. 2021;34(3):434–436. doi:10.1080/08998280.2021.1902727
51. Murillo-de-Ozores AR, Carbajal-Contreras H, Magana-Avila GR, et al. Multiple molecular mechanisms are involved in the activation of the kidney sodium-chloride cotransporter by hypokalemia. Kidney Int. 2022;102(5):1030–1041. doi:10.1016/j.kint.2022.06.027
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.